









Clostridium difficile has emerged as an increasingly important nosocomial pathogen and the prime causative agent of antibiotic-associated diarrhoea and pseudomembranous colitis in humans. In addition to toxins A and B, immunological studies using antisera from patients infected with C. difficile have shown that a number of other bacterial factors contribute to the pathogenesis, including surface proteins, which are responsible for adhesion, motility and other interactions with the human host. In this study, various clostridial targets, including FliC, FliD and cell wall protein 66, were expressed and purified. Phage antibody display yielded a large panel of specific recombinant antibodies, which were expressed, purified and characterised. Reactions of the recombinant antibodies with their targets were detected by enzyme-linked immunosorbent assay; and Western blotting suggested that linear rather than conformational epitopes were recognised. Binding of the recombinant antibodies to surface-layer proteins and their components showed strain specificity, with good recognition of proteins from C. difficile 630. However, no reaction was observed for strain R20291—a representative of the 027 ribotype. Binding of the recombinant antibodies to C. difficile M120 extracts indicated that a component of a surface-layer protein of this strain might possess immunoglobulin-binding activities. The recombinant antibodies against FliC and FliD proteins were able to inhibit bacterial motility.
Phage display technology was first described by George Smith,1 and since then it has emerged as an effective tool used in various biological fields. One of the most important applications is selection of recombinant antibodies, independent of the mammalian immune system. Based on the importance of antibodies in medical research and therapy2–4 and the need to move away from hybridoma technologies,5,6 libraries of recombinant molecules have emerged as key resources for antibody discovery.7–9
The bacterial pathogen Clostridium difficile is an anaerobic, Gram-positive, spore-forming organism, first discovered 80 years ago.10 It is the major cause of antibiotic-associated diarrhoeal disease and pseudomembranous colitis.11 Although the large proteins, toxins A and B, are well-characterised virulence factors; other molecules are likely to contribute to the disease process, notably those present on the bacterial surface. Cell surface proteins of C. difficile 630, such as Cwp66,12 Cwp8413 and SlpA,14 were analysed,15 and it has been shown that a significant number of them possess the cell wall-binding Pfam04122 motifs. Other surface proteins, including flagellar16 and GroEL-like proteins,17 which contribute to chaperone functions, may also directly or indirectly facilitate pathogenesis.
This study was aimed to isolate recombinant antibodies against certain surface proteins of C. difficile to facilitate characterisation of their location, function and contribution to the pathogenesis. A phage display library of humanised single-chain variable-fragment antibodies (scFvs) was screened against a range of recombinant clostridial proteins, and target-specific antibodies were characterised to elucidate their potential role in the biology of the pathogen.
Materials and methodsCulture of C. difficileThree strains of C. difficile (630, R20291 and M120) were obtained from a local culture collection. Autoclaved Brazier's CCEY agar (Oxoid, Hampshire, UK) supplemented with 10mL/L of cycloserine/cefoxitin (250/8mg/L) and 40mL/L of egg yolk was prepared for propagation of the organism. The bacterium was grown for 48h at 37°C under anaerobic conditions. Brain heart infusion (BHI) broth (20mL) was pre-incubated for 16h under anaerobic conditions, then inoculated with a single colony from an agar plate, and liquid cultures were grown under the above-mentioned conditions.
PCRAmplification of candidate sequences was performed in 50μL reactions, each containing 2μL of genomic DNA from C. difficile strain 630, 0.5μM each primer (Table 1), 200μM dNTPs and 0.5μL of Phusion polymerase (New England Biolabs, Ipswich, MA, USA). Thirty-five cycles of amplification were performed, each comprising denaturation at 94°C (30s), annealing at 55°C (30s) and extension at 72°C (1min). PCR products were characterised by electrophoresis in a 1% agarose gel and purified for cloning.
List of sense and antisense primers, with the predicted size and molecular weight of the targets with and without tag.
| Target name (CD) | Primers Sense (upper sequence) Antisense (lower sequence) | Predicted product size (BP) | Predicted molecular Mass of protein (kDa) | Predicted molecular mass including tag (kDa) |
|---|---|---|---|---|
| cwp84 (2787) | 5′ GAC GAC GAC AAG ATA GAT GGA GTA GAA ACT GCA GAG 3′ 5′ GA GGA GAA GCC CGG TTC ATT TCC ATT TCC ACC AAC 3′ | 2265 | 82.04 | 102.51 |
| cspA (0892) | 5′ GAC GAC GAC AAG ATG AAA AAC GGA ATA GTA AAA TGG 3′ 5′ GA GGA GAA GCC CGG TAC GTT TTC AGC TTG AGG TCC 3′ | 192 | 7.06 | 27.53 |
| 5′ cwp66 (2789) | 5′ GAC GAC GAC AAG ATA ACG GGT TCT GGA AGA TGG 3′ 5′ GA GGA GAA GCC CGG TTT AGC TGC TAA TAC ACC CAC 3′ | 744 | 26.6 | 47.07 |
| fbpA (2592) | 5′ GAC GAC GAC AAG ATA CAT CAA CCT GAA GAT GAT GAG 3′ 5′ GA GGA GAA GCC CGG TTT AAC CTT AAG CTT GGC TAC 3′ | 1689 | 64.71 | 85.18 |
| groEL (0194) | 5′ GAC GAC GAC AAG ATTGGA GTA ACT ATA GCA AAA GAG 3′ 5′ GA GGA GAA GCC CGG TCC GCC ACC CAT TCC TGG 3′ | 1461 | 51.73 | 72.2 |
| fliD (0237) | 5′ GAC GAC GAC AAG ATT CCA GTA AGA GTT ACA GGC 3′ 5′ GA GGA GAA GCC CGG TTG TGA GAA ATA GTT CAT TTG 3′ | 1497 | 55.31 | 75.78 |
| acd (1304) | 5′ GAC GAC GAC AAG ATT GAA CCA ACT GCC GAA AGT AGC 3′ 5′ GA GGA GAA GCC CGG TTC CAT AAT TCC AGA AAT TCC 3′ | 1650 | 59.75 | 80.22 |
| fliC (0239) | 5′ GAC GAC GAC AAG ATG GAG AAG TTA TCT TCT GGG 3′ 5′ GA GGA GAA GCC CGG TAA AAC TCC TTG TGG TTG TTG 3′ | 780 | 27.57 | 48.04 |
| 3′ cwp66 (2789) | 5′ GAC GAC GAC AAG ATA GTT ACT CAA ATT GGT GGC 3′ 5′ GAC GAC GAC AAG ATA GTT ACT CAA ATT GGT GGC 3′ | 900 | 33.47 | 53.94 |
| sortaseB (2718) | 5′ GAC GAC GAC AAG ATC AAT CAT GAT ACT AAA ATA TCC 3′ 5′ GA GGA GAA GCC CGG TCT ACCATGAATCAC C 3′ | 579 | 22.91 | 43.38 |
| cwp66 (2789) | 5′ GAC GAC GAC AAG ATA ACG GGT TCT GGA AGA TGG 3′ 5′ GAC GAC GAC AAG ATA GTT ACT CAA ATT GGT GGC 3′ | 1722 | 62.75 | 83.22 |
The nucleotides shown in bold letters are required for LIC in the expression vector.
Purified PCR products were cloned in the pET-32 Ek/LIC vector according to the manufacturer's instructions (Novagen, Madison, WI, USA). Aliquots (1μL) of the recombinant plasmid were transformed into thawed NovaBlue competent cells (50μL) using a brief heat shock at 40°C. Transformants were selected on L-agar containing ampicillin (50μg/mL), and then colony PCR and DNA sequencing were performed on the colonies selected. Plasmid DNA was purified from 2-mL cultures of the confirmed transformants and transformed into Escherichia coli BL21(DE3) and BL21(DE3) pLysS cells. Then, 2mL of a 16-h culture of the bacterial strain was added to 200mL of 2× YT–ampicillin (50μg/mL) medium and grown to an absorbance of 0.8 at 600nm. Isopropyl β-d-1-thiogalactopyranoside (IPTG, 1mM final concentration) was then added and the cultures were incubated for 16h at 30°C. Cells were collected from the 16-h cultures by centrifugation at 3500×g for 20min at 4°C, then resuspended in PBS and disintegrated by ultrasonication. Following centrifugation under the above conditions, recombinant histidine-tagged proteins were purified from the supernatants by nickel-chelate affinity chromatography on pre-packed 5-mL Hi-Trap columns (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Proteins were eluted with imidazole (100–500mM), and fractions were analysed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE), Western blotting and mass spectrometry.
Preparation of surface-layer (S-layer) extracts using low-pH glycine bufferCell wall proteins were extracted from cultures of the C. difficile strains (630, R20291 and M120) as described previously.15 Briefly, to extract S-layer proteins, bacterial pellets were resuspended in 0.04M glycine (pH 2.2), and after 30-min incubation at room temperature, intact cells were removed by centrifugation at 5000×g for 10min at room temperature. Supernatants containing the surface proteins extracted were neutralised to pH 7.0 with 2M Tris.
Expression of heat shock proteinsThe C. difficile strains (630, R20291 and M120) were grown in 20mL of BHI liquid medium for 16h under anaerobic conditions and then heat-shocked under the same conditions at 42°C for 2h. Bacterial pellets were separated at 5000×g for 10min at room temperature and stored at −20°C.
Selection of scFv antibodies by phage displayTomlinson libraries of humanised scFv antibodies (MRC Lab, Cambridge, UK) were used for isolation of antibodies against the recombinant clostridial target proteins. For selection, immunotubes (Nunc, Pasadena, TX, USA) were coated with a purified recombinant clostridial protein (4mL at a concentration of 100μg/mL in PBS). The tubes were washed three times with PBS, then blocked with 2% skim milk–PBS, and later 1013 phages from each library were added. After 2-h incubation, the unbound viruses were washed out 10 times with PBS containing 0.1% Tween 20, and the attached phages were recovered by the addition of trypsin (0.5mL, 1mg/mL). Half of the eluted phages were used to infect exponentially growing E. coli TG1. Serial dilutions of the infected bacteria were plated on tryptone-yeast extract agar, containing 100μg/mL of ampicillin and 1% glucose, to estimate the numbers of phages recovered by selection. The remaining infected bacteria were used for phage rescue by superinfection with 1010 plaque-forming units (PFUs) of the KM13 helper phage. Phage particles were precipitated from 16-h culture supernatants with 20% polyethylene glycol 6000–2.5M NaCl, collected by centrifugation at 3300×g for 15min and then titred on E. coli TG1 for the next round of selection.
Monoclonal phage enzyme-linked immunosorbent assay (ELISA)Individual colonies were selected from titration plates and grown in 96-well plates to assess the specificity of the scFv clones isolated by phage display. Each bacterial culture was superinfected with 109 PFUs of the KM13 helper phage to produce clonal virus stocks. After overnight growth, the plates were centrifuged at 1800×g for 10min, and the supernatants were used in ELISA on plates pre-coated with relevant recombinant clostridial proteins. Phage binding was detected with a monoclonal anti-M13 antibody conjugated to horseradish peroxidise (Sigma, Dorset, UK).
Expression and purification of soluble scFv antibodiesPhages with properties of interest were used to infect non-suppressor E. coli strain HB2151, which was plated on selective agar containing 100μg/mL of ampicillin. The transfected bacteria were then grown in a liquid medium to the late exponential phase (absorbance of 0.9 at 600nm) before addition of IPTG to a final concentration of 1mM. Growth was continued for 16h at 30°C, and then the culture supernatants were used in ELISA using a protein A–HRP conjugate to detect scFv binding to the relevant recombinant clostridial protein. The scFvs were precipitated from the culture supernatants using ammonium sulphate and dialysed against PBS, pH=7. The dialysed samples were purified by nickel affinity chromatography and analysed by Western blotting using an anti-c-myc antibody and an anti-rabbit-HRP conjugate (Sigma, Dorset, UK) to detect the scFvs. Surface-layer protein A (SlpA) was extracted from C. difficile 630, separated by SDS–PAGE and then transferred to a Hybond membrane. The membrane was probed with individual scFvs to examine their binding by Western blotting.
Motility inhibition testBacterial motility inhibition assays were performed to evaluate the properties of recombinant antibodies against flagellar proteins. Cells of C. difficile strain 630 were serially diluted, and 105 bacterial cells were incubated with 150μL of the purified scFvs against single protein components of the flagellar structure, along with control reactions including M120 as a non-motile strain. The mixtures were incubated for 1h at 37°C under anaerobic conditions, and then one-tenth of the mixture was deeply inoculated into 0.2% BHI agar. The cultures were kept under the above-mentioned conditions and monitored for growth over 48–72h.
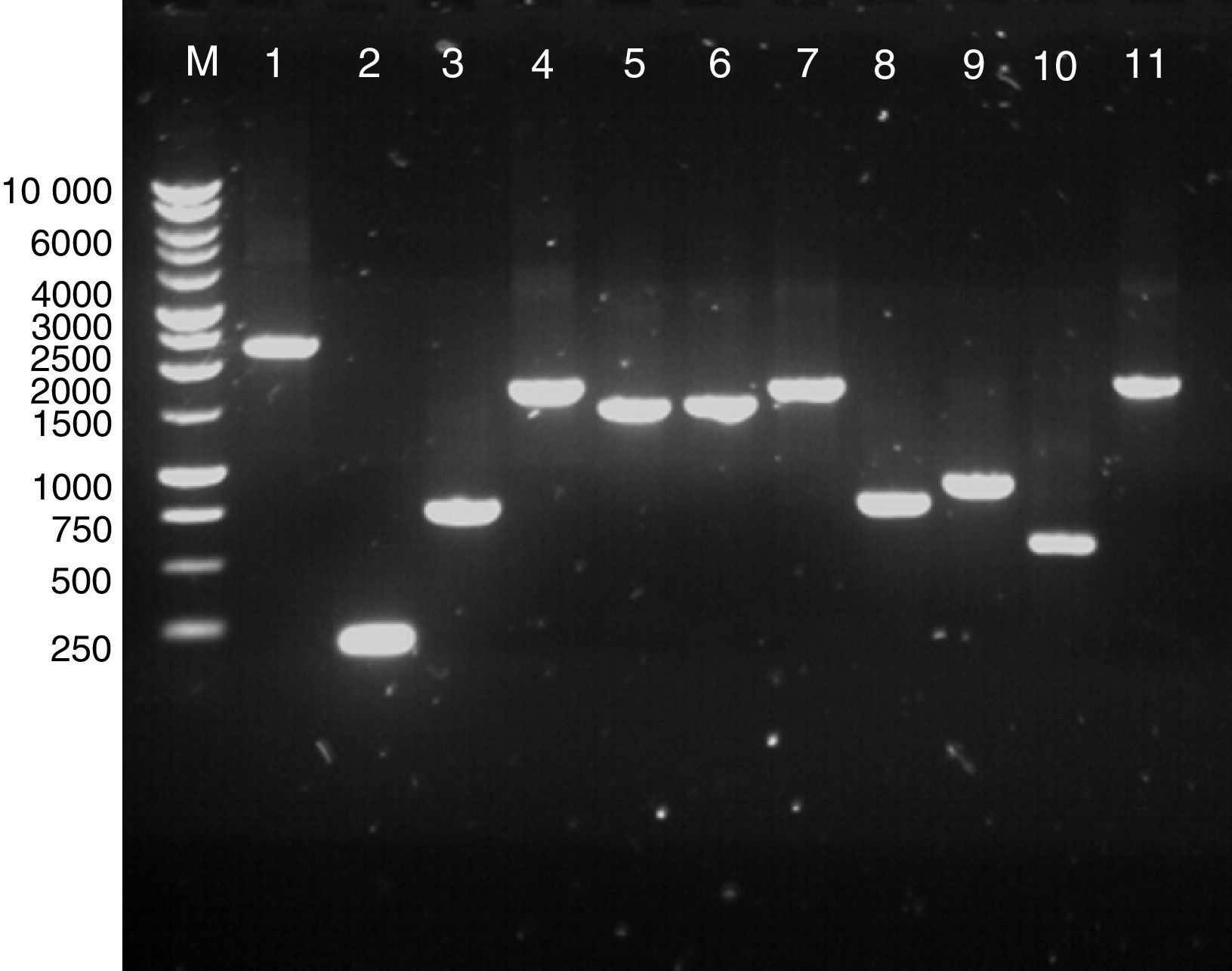
ResultsPCR of clostridial targetsAmplification of the genes for a number of clostridial proteins with a putative surface location was optimised using gene-specific primers (Table 1). The targets included Cwp84, Cwp66, Acd, GroEL, FliD, FliC, CspA, FbpA and a putative sortase. The amplicons were analysed by gel electrophoresis, their sizes were confirmed as predicted from the genomic sequence of C. difficile 630 (Fig. 1), and the DNA was isolated for cloning.
; lane 2, cspA (189bp); lane 3, 5′ region of cwp66 (744bp); lane 4, fbpA (1689bp); lane 5, groEL (1461); lane 6, fliD (1497bp); lane 7, acd (1650bp); lane 8, fliC (780bp); lane 9, 3′ region of cwp66 (900bp); lane 10, sortaseB (579bp); lane 11, cwp66 (1722bp). All PCR reactions were carried out with Phusion DNA polymerase with buffer HF (lanes 2, 3, 5, 6, 7, 8, 9, 10) or GC buffer (for lanes 1, 4 and 11).")
Agarose gel electrophoresis of extracted PCR products. Lane M, 1kb DNA ladder; lane 1, cwp84 (predicted size, 2265bp); lane 2, cspA (189bp); lane 3, 5′ region of cwp66 (744bp); lane 4, fbpA (1689bp); lane 5, groEL (1461); lane 6, fliD (1497bp); lane 7, acd (1650bp); lane 8, fliC (780bp); lane 9, 3′ region of cwp66 (900bp); lane 10, sortaseB (579bp); lane 11, cwp66 (1722bp). All PCR reactions were carried out with Phusion DNA polymerase with buffer HF (lanes 2, 3, 5, 6, 7, 8, 9, 10) or GC buffer (for lanes 1, 4 and 11).
Colony PCR analysis using the target-specific primers showed that most of the transformants carried inserts of the size expected for each target, which were further confirmed by sequencing (S-tag or T7-specific primers, Novagen, Madison, WI, USA). Except cwp84, all the inserts were of the right length and in a correct frame with the fused tag used for detection and purification of the recombinant proteins. In the case of cwp84, original amplification appeared successful (Fig. 1, lane 1); however, deletion most likely occurred during ligation or transformation. When the E. coli DE3 bacterial transformants were induced, Western blotting of the lysates showed successful expression of most clostridial target proteins in the E. coli background, and proteins of predicted molecular weights were detected on the blots (Fig. 2). Peptidoglycan hydrolase Acd was the only exception, for which no signal was detected (Fig. 2, lane 1).
 cells expressing the five target proteins. Lane M, SeeBlue Plus2 pre-stained molecular weight markers (Invitrogen); lane 1, Acd; lane 2, FliC; lane 3, C-terminus of Cwp66; lane 4, SortaseB; lane 5, Cwp66. After transfer and blocking, the recombinant proteins were detected with anti S-tag antibody, anti mouse-HRP and TMB substrate.")
Western blot analysis of the extracts of E. coli BL21 (DE3) cells expressing the five target proteins. Lane M, SeeBlue Plus2 pre-stained molecular weight markers (Invitrogen); lane 1, Acd; lane 2, FliC; lane 3, C-terminus of Cwp66; lane 4, SortaseB; lane 5, Cwp66. After transfer and blocking, the recombinant proteins were detected with anti S-tag antibody, anti mouse-HRP and TMB substrate.
Most of the purified recombinant targets were identical with the proteins of C. difficile 630, as confirmed by SDS–PAGE, mass spectrometry and Mascot analysis (data not shown). These proteins were sortase B, CspA, GroEL, FliC and FliD (flagellar components), N- and C-terminals of Cwp66 along with the complete reading frame of Cwp66. A low-molecular-weight form of SlpA (designated LMW) and a complete surface-layer structure comprised of high- and low-molecular-weight components (designated SlpA) were obtained from our collaborators. These materials were used as targets in phage display to recover specific recombinant scFv antibodies.
Panning and selection of antibody phages from scFv libraries I and JAnalysis performed during three rounds of selection revealed increases in phage recovery rates. The monoclonal phage ELISA assays demonstrated that 50–100% of all clones were reactive against the intended target, although there was some variation in the ELISA signal measured by absorbance at 450nm (data not shown).
Expression and purification of soluble scFvs against clostridial targetsThe scFvs producing the best signals in the ELISA assays against the targets were scaled up using high-volume cultures. Supernatants from the induced E. coli HB2151 transfectants were purified by nickel affinity chromatography and using the tag encoded by the display/expression vector, fused at the termini of the recombinant antibodies. High yields of each culture were obtained, and a series of 30-kDa proteins was purified, predicted to be scFvs (Fig. 3). In general, the yields were consistent, and 30-kDa proteins were observed in all instances, despite the fact that one recombinant antibody (anti-LMW scFv clone G7; lane 2) was present at high yield and another was only weakly visible in the gel (anti-LMW scFv clone A6; lane 4).
 and extracted SlpA (lanes 7–9). Columns were loaded with the sample after adjusting pH and buffer conditions and washed with binding buffer initially without imidazole and then with buffer containing a low concentration of imidazole (40mM). The scFv protein was eluted with 200mM imidazole in binding buffer. Lane M, SeeBlue Plus2 Pre-Stained molecular weight markers (Invitrogen); lane 1, anti-LMW clone F10; lane 2, G7; lane 3, D4; lane 4, A6; lane 5, G1; lane 6, H1; lane 7, anti-SlpA clone E10; lane 8, A10; lane 9, A9.")
SDS-PAGE analysis of scFvs against recombinant LMW (lanes 1–6) and extracted SlpA (lanes 7–9). Columns were loaded with the sample after adjusting pH and buffer conditions and washed with binding buffer initially without imidazole and then with buffer containing a low concentration of imidazole (40mM). The scFv protein was eluted with 200mM imidazole in binding buffer. Lane M, SeeBlue Plus2 Pre-Stained molecular weight markers (Invitrogen); lane 1, anti-LMW clone F10; lane 2, G7; lane 3, D4; lane 4, A6; lane 5, G1; lane 6, H1; lane 7, anti-SlpA clone E10; lane 8, A10; lane 9, A9.
Approximately 600 clones with a high binding capacity to the clostridial target proteins were identified, and of these 30 scFvs were chosen for sequence analysis. Plasmid DNAs were purified and sequenced using primers for flanking vector sequences and the primers that annealed to the linker coding sequence separating the heavy- and light-chain domains of each scFv. The results showed that the framework sequences were identical, as expected from the manner in which the libraries were constructed. Amino acids present at diversified positions in the complementarity determining regions, CDR2 and CDR3, of the heavy- and light-chains are assembled for comparison in Table 2. In some cases, all scFv antibodies against a particular clostridial target were unique, such as clones A9, E7 and G12 directed against CspA (Table 2). The diversified residues in these antibodies were all distinctive, although E7 and G12 contained threonine or serine residues in the heavy-chain domain with certain frequencies. In other groups, near-identical sequences were recovered by screening of the phage display libraries against a defined target. Similar properties were also evident among the scFvs against Cwp66 and sortase B (Table 2). The six clones against LMW were as follows: A6 and D4 were identical, G1 and H1 formed a second identical pair, whilst F10 and G7 were unique. The results of restriction enzyme (NcoI and NotI) analysis conducted to evaluate the presence of full-length scFv reading frames showed that all the scFvs were intact (Fig. 4).
Amino acids present in CDR2 and CDR3 in heavy and light chains of sequenced clones. Columns designate the position in the chains reading frame (e.g. “H50”, amino acid residue 50 in the scFv heavy chain; “L50”, amino acid residue 50 in the scFv light chain; Kabat numbering system). Rows identify the clostridial target and the clone chosen for sequence analysis. “G/E” designates a residue that could not be resolved from sequence traces.
| CLONES | H50 | H52 | H52a | H53 | H55 | H56 | H58 | H95 | H96 | H97 | H98 | L50 | L53 | L91 | L92 | L93 | L94 | L96 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CspA A9 | T | E | K | Q | E | S | D | R | K | P | P | R | H | R | M | R | A | L |
| CspA E7 | T | T | T | P | A | T | T | T | R | R | M | H | R | G | T | T | L | M |
| CspA G12 | S | S | R | T | K | G | F | L | S | K | R | A | S | Q | H | R | A | H |
| N-terminus of Cwp66 A1 | A | G | Y | S | S | A | R | N | A | Y | T | A | Y | S | D | N | S | A |
| N-terminus of Cwp66 F4 | T | S | R | L | T | N | L | N | G | A | L | A | L | S | S | V | S | P |
| N-terminus of Cwp66 H2 | S | G | Y | S | S | A | I | N | A | Y | T | A | Y | S | D | N | S | A |
| C-terminus of Cwp66 A11 | G | N | Y | D | Y | T | S | Y | S | T | S | H | G | T | A | D | S | S |
| C-terminus of Cwp66 B9 | G | T | A | N | Y | T | S | T | G | S | N | N | S | D | G | N | S | D |
| C-terminus of Cwp66 C9 | R | I | P | N | Y | T | E | S | G | H | T | N | I | N | S | D | A | F |
| Cwp66 A5 | T | S | R | L | T | N | L | N | G | A | L | G | S | A | W | A | L | N |
| Cwp66 C1 | T | S | R | L | T | N | L | N | G | A | L | A | L | S | S | V | S | P |
| Cwp66 F4 | S | S | R | L | T | N | L | N | G | A | L | A | L | S | S | V | S | P |
| GroEL E10 | H | T | K | E | T | G | W | R | R | H | H | G | Y | F | S | R | K | S |
| FliC A4 | G | N | S | N | Y | T | S | Y | G | S | N | D | N | S | N | S | Y | S |
| FliC B5 | G | T | A | N | Y | T | S | S | S | T | S | N | Y | G | N | N | N | S |
| FliC C9 | R | I | P | N | T | T | E | S | G | H | T | Q | N | P | V | S | A | P |
| FliD B4 | S | T | S | T | D | Y | G | T | S | S | N | G | S | D | S | N | T | A |
| FliD F6 | S | A | Y | G | D | Y | S | S | A | Y | N | S | A | A | S | N | T | G |
| SortaseB B8 | T | T | S | L | D | F | S | P | A | A | T | A | S | T | T | S | T | D |
| SortaseB D10 | S | T | S | L | D | F | S | P | A | A | T | A | S | N | L | S | T | D |
| SortaseB D11 | S | T | S | L | D | F | Q | P | S | A | T | A | S | N | T | S | T | D |
| LMW A6 | S | S | G | T | Y | S | A | G | D | S | F | S | T | A | S | S | S | T |
| LMW D4 | S | S | G | T | Y | S | A | G | D | S | F | S | T | A | S | S | S | T |
| LMW F10 | S | Y | A | ? | T | R | K | H | P | L | I | M | M | K | K | A | T | A |
| LMW G1 | T | G | T | Y | S | S | A | N | A | A | A | A | S | Y | A | Y | Y | T |
| LMW G7 | G | S | T | H | S | R | Q | N | G | T | L | Q | E | A | Q | S | N | Q |
| LMW H1 | T | G | T | Y | S | S | A | N | A | A | A | A | S | Y | A | Y | Y | T |
| SlpA A9 | G | S | T | H | S | R | Q | N | G | T | L | Q | E | A | Q | S | N | Q |
| SlpA A10 | T | S | T | A | K | G | H | N | Y | P | A | P | N | H | A | T | T | P |
| SlpA E10 | L | N | A | A | T | R | L | S | M | R | A | K | R | G/E | M | S | G | T |
; lane 1, FliD B4; lane 2, FliD B5; lane 3, FliD A6; lane 4, FliC B5; lane 5, FliC F1; lane 6, LMW F10; lane 7, LMW H1; lane 8, LMW G7; lane 9, LMW G1; lane 10, LMW A6; lane 11, LMW D4; lane 12, SlpA A10; lane 13, SlpA E10; lane 14, SlpA A9.")
Restriction analysis for the presence of full-length scFv inserts in clones from the Tomlinson library. Plasmid DNA from each clone was digested with NcoI and NotI. Release of a full-length scFv insert is expected to result in a fragment of 717bp. Lane M, 1kb DNA ladder (Promega); lane 1, FliD B4; lane 2, FliD B5; lane 3, FliD A6; lane 4, FliC B5; lane 5, FliC F1; lane 6, LMW F10; lane 7, LMW H1; lane 8, LMW G7; lane 9, LMW G1; lane 10, LMW A6; lane 11, LMW D4; lane 12, SlpA A10; lane 13, SlpA E10; lane 14, SlpA A9.
All purified scFvs were able to recognise the recombinant clostridial targets used in selection by Western blots (data not shown). Given that recombinant methods were used for production of these proteins in E. coli, analysis was carried out to study the recognition of native forms of each protein.
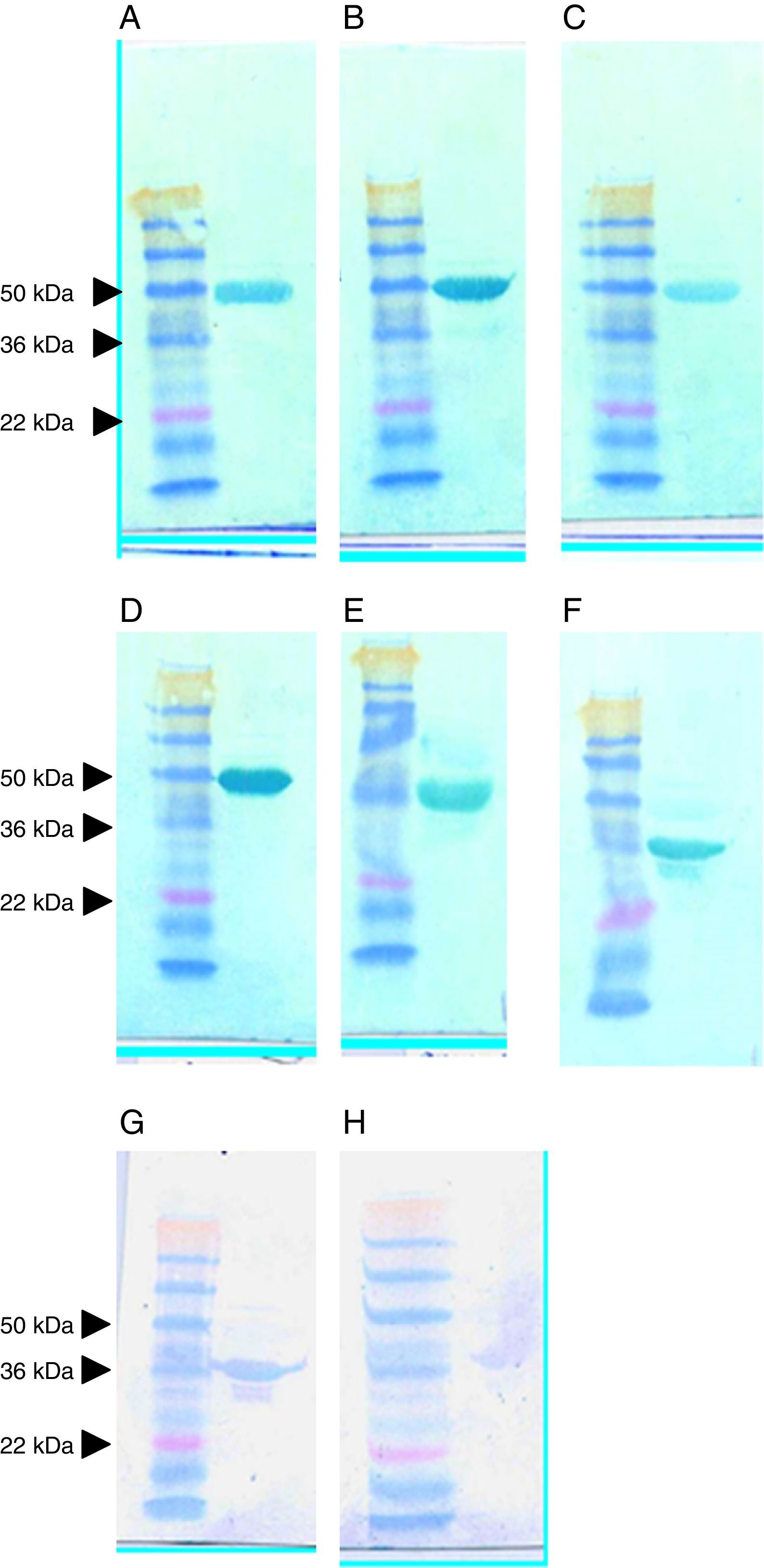
All the anti-SlpA scFvs recognised the native target protein from C. difficile 630 on a Western blot; however, the patterns of recognition were sometimes unexpected. As shown in Fig. 5, anti-LMW scFvs D4, H1, A6 (upper panel, A–C, respectively), G1 and F10 (middle panel, D and E, respectively) all recognised the high-molecular-weight (HMW) component of the native SlpA complex, despite having been isolated by selection on a recombinant form of the LMW protein. The recognition was strong, specific and unambiguous. In contrast, G7 (Fig. 5, middle panel, F) isolated in the same selection experiment reacted strongly with the LMW protein. This result was also noticeable in the reactions of SlpA A9 and SlpA E10; meanwhile, these scFvs were isolated by selection on the native SlpA complex.
. Upper panel: A, LMW scFvs D4; B, H1; C, A6. Middle panel: D, LMW G1; E, F10; F, G7. Bottom panel: G, SlpA A9; H, E10.")
Western blot analysis of the reaction of scFvs with SlpA extracted from C. difficile 630. In each panel, the left hand lane shows the migration of SeeBlue Plus2 pre-stained molecular weight markers (Invitrogen). Upper panel: A, LMW scFvs D4; B, H1; C, A6. Middle panel: D, LMW G1; E, F10; F, G7. Bottom panel: G, SlpA A9; H, E10.
The scFvs against the SlpA protein of C. difficile 630 were also analysed on Western blots using extracts from C. difficile 630, R20291 and M120. None of the scFvs were able to bind to the SlpA components of C. difficile R20291, but all showed ability to attach to the SlpA components of strains 630 and M120 (data not shown). When analysing the extracts from C. difficile 630, R20291 and M120, reactions between the scFvs and the putative native targets were generally obvious. However, it was consistently observed, regardless of the selection or specificity, that the antibodies were bound to a protein from M120, which was of a similar molecular weight to that of the LMW component of SlpA (Fig. 6). In these experiments, extracts from C. difficile 630, M120 and R20291 were probed with three scFv clones against GroEL, of which only clone E10 could react with its native target (58kDa). However, a weak signal was also evident at a lower molecular weight in the extract from strain 630, and a much weaker signal was evident in the extract from strain R202091, but the strongest signal was detected at 36kDa (Fig. 6, unshaded arrowhead). This suggests a reaction between scFv E10 and the LMW component of SlpA from the M120 strain. This finding was consistent when extracts of M120 were probed, irrespective of the specificity of the scFv and its reactivity with components in extracts from M120 and the other strains, suggesting the presence of an immunoglobulin-binding activity in extracts from C. difficile strain M120.
. Anti c-myc and anti-rabbit-HRP conjugate were used to detect the binding of scFv. The left hand lane shows the migration of SeeBlue Plus2 pre-stained molecular weight standards (Invitrogen). Moving to right, the lanes show extracts from C. difficile 630, M120 and R20291 respectively.")
Western blot analysis of reaction of scFv with GroEL extracted from C. difficile 630, M120 and R20291. Heat shocked bacterial lysate was prepared from the three strains, separated by SDS-PAGE and transferred to Hybond membrane that was then probed with anti-GroEL scFv (from left E10, A1 and E9). Anti c-myc and anti-rabbit-HRP conjugate were used to detect the binding of scFv. The left hand lane shows the migration of SeeBlue Plus2 pre-stained molecular weight standards (Invitrogen). Moving to right, the lanes show extracts from C. difficile 630, M120 and R20291 respectively.
The scFvs against the FliD (clone B5) and FliC (clone A6) proteins were tested to assess whether their binding to the bacterial surface would interfere with a specific biological activity such as motility (Fig. 7). In the images, bacterial mobility is apparent as diffuse growth from the stab inoculation site. The three left images in the top row show the growth of C. difficile 630 without an scFv (left) and pre-mixed with an irrelevant scFv prior to inoculation (middle), as well as the growth of the non-motile M120 strain without an scFv (right). These images illustrate the diffuse pattern of growth of motile bacteria. The three right images in the top row of Fig. 7 show the growth of C. difficile 630 pre-incubated with undiluted (left), 10-fold diluted (middle) and 100-fold diluted (right) anti-FliC scFv B5. The images in the bottom row of Fig. 7 show the results obtained using the same experimental design with anti-FliD scFv A6. Although inhibition of bacterial motility by the anti-FliC and anti-FliD scFvs could be seen under all conditions, the range of inhibition gradually decreased with the increase of the dilution factor.
, scFv of irrelevant specificity (centre) and the non-motile strain M120 without scFv (right). Upper right pannel: C. difficile 630 pre-incubated with anti-FliC scFv B5 (undiluted: left; 1/10: centre; 1/100: right). Bottom row: C. difficile 630 pre-incubated with anti-FliD scFv A6 (undiluted: left; 1/10: centre; 1/100: right).")
Effect of recombinant antibodies on the motility of C. difficile. Bacteria were mixed with scFvs, incubated, then stabbed to agar and cultured for 48h. Upper left pannel: C. difficile 630 without scFv (left), scFv of irrelevant specificity (centre) and the non-motile strain M120 without scFv (right). Upper right pannel: C. difficile 630 pre-incubated with anti-FliC scFv B5 (undiluted: left; 1/10: centre; 1/100: right). Bottom row: C. difficile 630 pre-incubated with anti-FliD scFv A6 (undiluted: left; 1/10: centre; 1/100: right).
In many intracellular and extracellular bacterial pathogens, surface proteins play a crucial role in the pathogenesis.18,19 Synthesis of these proteins, their assembly on the surface, activities and strain-to-strain differences are all promising areas for exploitation in the development of therapeutics and diagnostics. Analysis of the C. difficile proteome indicates that approximately 50 surface components exist, many being SlpA paralogs and others having distinctive functions such as a role in bacterial motility.
Phage display technology has provided a fast, efficient and productive strategy for isolation of monoclonal antibodies and allowed the screening and analysis of significant numbers of clones. In this study, from the initial panel of 600 soluble antibodies, 30 were chosen for the final panel of reagents. This is the first report of the recombinant antibody selection against a range of surface proteins of C. difficile.
Besides the unexpected interactions of selected anti-SlpA 630 antibodies with low- and high-molecular-weight components, analysis of these binders against SlpA revealed that the antibodies bind to extracts of the C. difficile 630 and M120 strains under both denaturing and non-denaturing conditions; however, the scFvs failed to bind to extracts from the R2029 strain. This suggests that the epitopes for this panel of antibodies were probably linear, and hence treatment with SDS did not disrupt the interaction sites. SlpA is a major protein of C. difficile,20 comprised of two parts, of which the HMW component may have a role in binding of the bacterium to host proteins, such as collagen. Accordingly, the potential exists for bacterial adhesion to be inhibited by specific immunological reagents.21 Regarding the anti-SlpA scFvs isolated in the current work, one future opportunity would be to look for anti-HMW antibodies and test if they possess similar properties. One of the targets was GroEL as a heat shock protein. Among all the scFvs selected against recombinant clones of this protein, only clone E10 could react with the native form. The results obtained with the extracts of the C. difficile M120 strain showed that most of the scFvs used to probe Western blots appeared to be able to bind to a protein of about 36kDa, a size consistent with that of the LMW component of SlpA from the M120 strain. Reactions with components of this size were evident by analysing the scFvs directed against GroEL, sortase B, FliC and CspA (data not shown). Given the number of the scFvs that appeared to react with this protein, the most likely explanation is that it is able to bind scFvs not through the interaction with residues in the CDRs of the antibodies, but via other parts of the recombinant immunoglobulin, similar to the cases of A and L proteins. It has also been reported that some spore proteins of Bacillus subtilis are able to interact with scFvs, indicating that there exist certain VH variants of the antibodies.22
The current study showed that binding of scFvs to the FliC and FliD targets could partially inhibit bacterial motility. Alignment of the FliC and FliD protein sequences from two strains (630 and 027) showed a high degree of identity between the two proteins, with greater variation in their central regions. This alignment and the blotting results suggest that the binding site for the anti-FliC scFvs may lie in the variable central regions (data not shown). It has been reported that FliC from six strains reacted with a polyclonal anti-FliC antibody23; however, the molecular weight of the protein, predicted from the gene sequence, was different from that of FliC isolated from the bacterial extracts. More recent bioinformatics analysis of different strains has shown that the protein undergoes glycosylation, and differences in the glycan biosynthesis genes further affect the observed masses of FliC from different strains.24
In conclusion, this study validated a strategy for generating recombinant antibodies against selected targets from the bacterial pathogen C. difficile. These reagents have a potential in diagnostics, as well as for advancing understanding of the disease process and developing new therapeutics.
Conflicts of interestThe authors declare no conflicts of interest.
We gratefully acknowledge the expert assistance of the staff of the Glasgow Biomedical Research Centre. Ali Nazari Shirvan wishes to thank his sponsors, the Ministry of Health, Iran, and the Razi Vaccine Institute for their support for this work.