





El síndrome hemolítico urémico atípico es una variante de la microangiopatía trombótica, caracterizado por una excesiva activación del complemento. Se caracteriza por presentar anemia hemolítica no autoinmune, trombocitopenia y falla renal aguda. Se ha observado que el 60% de los pacientes presentan mutaciones en los genes que codifican al complemento, tanto reguladores (factor H, factor I, cofactor de proteínas de membrana y trombomodulina), activadores (factor B y C3) como autoanticuerpos contra el factor H. Se requiere la presencia de múltiples factores para su manifestación, estos incluyen: un disparador y mutaciones de genes con la penetrancia adecuada. Es necesario que el clínico esté familiarizado con la enfermedad, ya que presenta una elevada morbimortalidad que puede ser modificada si se identifica de manera temprana y se da un tratamiento oportuno.
Atypical haemolytic uraemic syndrome is one of the main variants of thrombotic microangiopathy, and is characterized by excessive complement activation in the microvasculature. It is also characterised by the clinical triad; non-immune haemolytic anaemia, thrombocytopenia, and acute renal failure. In addition, 60% of patients have mutations in the genes encoding complement regulators (factor H, factor I, membrane cofactor proteins, and thrombomodulin), activators (factor B and C3), as well as autoantibodies against factor H. Multiple factors are required for the disease to manifest itself, including a trigger and gene mutations with adequate penetration. Being one of the differential diagnoses of preeclampsia- eclampsia and HELLP syndrome means that the clinician must be familiar with the disease due to its high mortality, which can be modified with early diagnosis and comprehensive treatment.
El síndrome hemolítico urémico atípico es una variante de la microangiopatía trombótica que se caracteriza por la tríada: anemia hemolítica no autoinmune, trombocitopenia y falla renal aguda1,2.
Se caracteriza por una irregularidad del complemento, ocasionada por una mutación genética de sus inhibidores2. La hipertensión maligna, la septicemia, los desórdenes autoinmunes (lupus, esclerodermia), las infecciones estreptocóccicas, el embarazo, el síndrome de HELLP y el cáncer pueden ser causantes de este síndrome3.
Tradicionalmente, el síndrome hemolítico urémico se clasifica en 2 formas: el síndrome urémico hemolítico típico, que tiene su pico de incidencia en la población infantil y es ocasionado por infecciones entéricas secundarias a bacterias productoras de toxina Shiga (fig. 1) y el síndrome urémico hemolítico atípico, que se asocia en el 50-60% de las veces a pacientes con mutaciones de genes dentro del sistema del complemento, que causa en la mayoría de los afectados, una insuficiencia renal crónica terminal y la necesidad de trasplante renal4–6.

El embarazo puede ser un disparador de esta enfermedad, en especial durante el puerperio. Esto es debido a que el complemento tiene un rol importante en la fisiopatología del embarazo, este se incrementa para prevenir el daño ocasionado por la placenta mediante la expresión trofoblástica de los reguladores del complemento, conocidos como factor acelerador de la degradación, proteína cofactor de membrana (PCM) y CD595-7.
En el puerperio existe una disminución de estas proteínas, o una reducción de la mayoría de las proteínas del complemento, que dan paso a la manifestación de la enfermedad7.
EpidemiologíaLa microangiopatía trombótica asociada al embarazo (P-TMA) tiene una incidencia de 1 de cada 250,000 embarazos8-10. Existen reportes de una incidencia de síndrome hemolítico urémico atípico de 2 casos por cada 1,000,000 de habitantes11. En mujeres en edad reproductiva el fenotipo de síndrome hemolítico urémico atípico se presenta en el embarazo tardío o en el puerperio inmediato11. En el 10% de las pacientes con este síndrome, el embarazo será el disparador12.
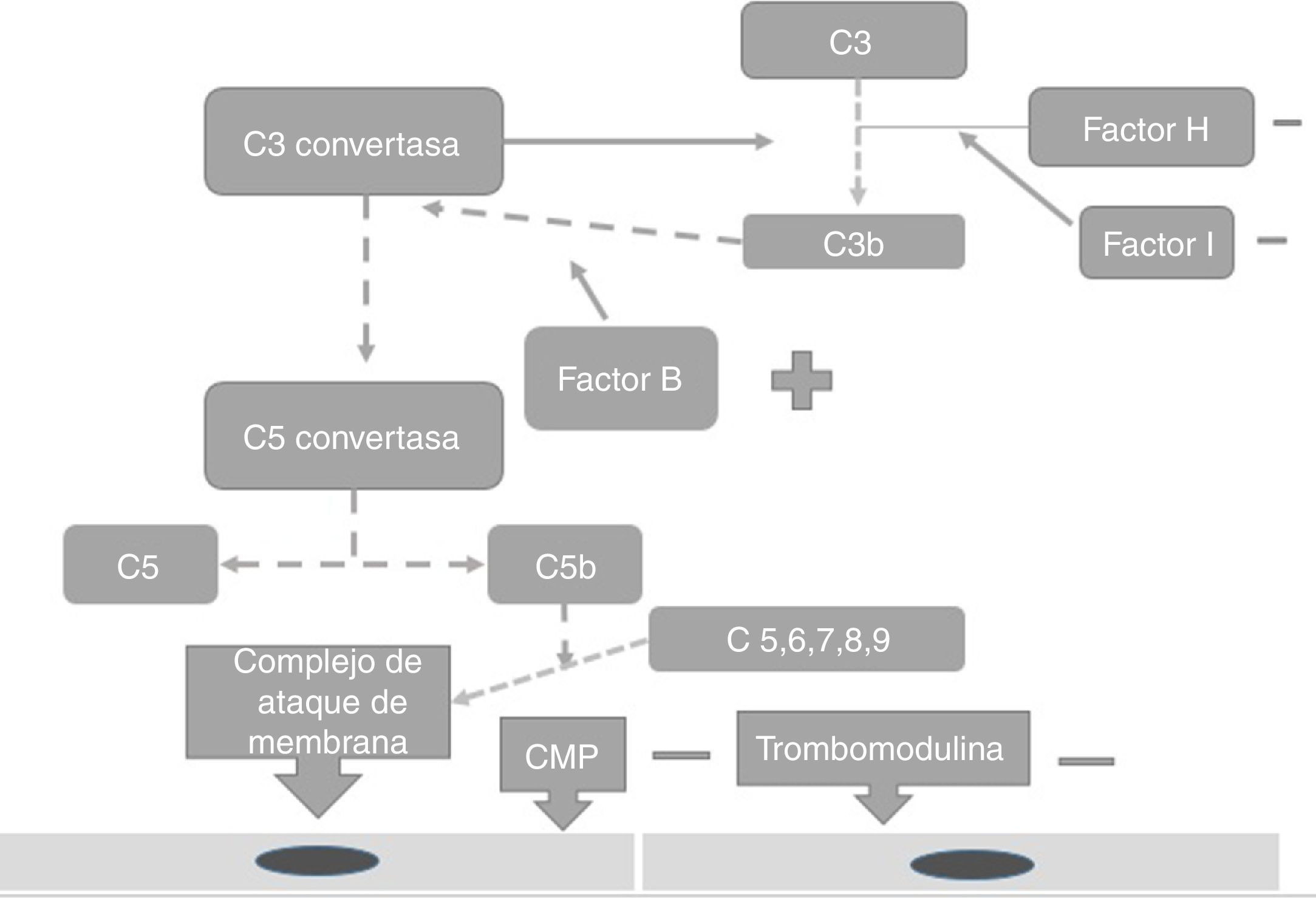
FisiopatologíaEl complemento es uno de los principales mecanismos efectores de la inmunidad mediada por anticuerpos, que se considera un puente entre la inmunidad innata y la adaptativa, que ofrece: protección contra la infección bacteriana, favorece la eliminación de complejos inmunes y de productos de la inflamación, ofrece protección contra agentes externos y regula la apoptosis celular13. Existen 3 vías independientes para su activación: la clásica, la de la lectina y la alternativa14. Su activación necesita estar regulada para prevenir el daño tisular, en especial cuando se activa la vía alternativa15. En la mayoría de las pacientes afectadas por síndrome hemolítico urémico atípico, el desarrollo de este se encuentra relacionado con la activación descontrolada de la vía del complemento y con un número creciente de mutaciones genéticas que ya han sido identificadas para la activación de este síndrome15. Estas mutaciones se observan en los genes que regulan la función del complemento, como los inhibidores del factor del complemento H (FCH). Richard16 identificó la importancia de las mutaciones de este gen como causa de síndrome hemolítico urémico, encontró mutaciones del gen FCH en los exones 18-20 de 2 familiares y de 3 pacientes esporádicos de los 19 familiares y en 31 pacientes esporádicos estudiados15. Por otra parte, este estudio demostró que el síndrome hemolítico urémico familiar es una condición heterogénea. Rodríguez17 encontró que las mutaciones en los reguladores FCH, factor de complemento I (FCI) y PCM ocasionan la pérdida de la función del complemento, mientras que los de C3 activan su funcionamiento (fig. 2). La PCM (CD46) es un complemento regulador transmembrana ampliamente expresado como el FCH, y Richards18 encontró una mutación de PCM (CD46) en individuos de 3 familias, los cuales presentaban una deleción de 2 aminoácidos (D237/S238) en la familia 1 (heterocigoto) y una sustitución S206P, en la familia 2 (heterocigoto) y 3 (homocigoto), con lo que comprobó que la desregulación del complemento predispone al desarrollo de microangiopatía trombótica y que las pacientes de la revisión para tales defectos podrían proporcionar estrategias preventivas de tratamiento en pacientes con este tipo de mutaciones18. La mutación del FCH ocurre en el 6-20% de las pacientes con síndrome hemolítico urémico atípico y la mutación en CD46 en el 10% de las pacientes18-22.

El factor I es un inhibidor de todas las vías del complemento, tiene la habilidad de degradar proteínas activadas C3b y C4b en presencia de cofactores como el FCH, CD46, entre otros. La deficiencia incompleta del factor I también está asociada al desarrollo del síndrome hemolítico urémico atípico19.
También el incremento de función de los factores del complemento, como los factores B y C3, pueden causar la expresión de este síndrome20-22. Autoanticuerpos en contra del FCH incrementan el riesgo para la expresión de este, sin embargo, la enfermedad tiene una penetrancia incompleta asociada de forma diferente a cada mutación y por lo tanto, no todas las portadoras de mutaciones desarrollarán síndrome hemolítico urémico atípico22,23 solo aquellas mujeres con predisposición genética y un disparador externo24.
Síndrome hemolítico urémico atípico y embarazoLa patogénesis del síndrome hemolítico urémico atípico en el embarazo permanece incierta, aparece en el 21% de las mujeres adultas con síndrome hemolítico urémico atípico y el 79% se manifiesta en el posparto.
El riesgo de presentar síndrome urémico hemolítico atípico gestacional es mayor durante el segundo embarazo. Fakhouri25, en un estudio retrospectivo, encontró anormalidades del complemento en 18 de 21 pacientes. Los resultados no difirieren entre las pacientes con embarazo relacionado y no relacionado con síndrome hemolítico urémico atípico. Las mutaciones en los dominios SCR19-20 de factor H fueron menos frecuentes en pacientes con síndrome hemolítico urémico atípico gestacional en comparación con al síndrome hemolítico urémico atípico. Los embarazos con anomalías del complemento presentaron complicaciones caracterizadas por pérdidas fetales y preeclampsia en el 4.8 y el 7.7%, respectivamente.
Diagnóstico diferencialLa microangiopatía puede ser manifestación de múltiples enfermedades, como: enfermedades del tejido conectivo, cáncer o en pacientes postrasplantados. Sin embargo, es un hallazgo dominante en la púrpura trombocitopénica trombótica, el síndrome hemolítico urémico atípico y el síndrome hemolítico urémico causado por toxina Shiga de Escherichia coli. La púrpura trombocitopénica trombótica está asociada a una deficiencia de ADAMTS 13, y ambos tipos de síndrome urémico están caracterizados por la presencia de anemia hemolítica, trombocitopenia y falla orgánica multiple26; el síndrome hemolítico urémico atípico se presenta durante el puerperio mientras que la púrpura trombocitopénica trombótica asociada a deficiencia de ADAMTS 13 aparece durante el embarazo26.
Un punto clave para diferenciar el síndrome hemolítico urémico atípico de la púrpura trombocitopénica trombótica es evidenciar la presencia de una deficiencia severa de ADAMTS 13, la cual debe ser mayor del 5-10%26-28. En los casos en los que no se puede estimar la actividad de ADAMTS 13, Coppo demostró que la presencia de un recuento plaquetario de 30×109/l y una creatinina por debajo de 2.26mg/dl se asocia a la deficiencia de ADAMTS 13 y por lo tanto, al diagnóstico de púrpura trombocitopénica trombótica (tabla 1)29.
Diagnósticos diferenciales del síndrome hemolítico urémico atípico y otras microangiopatías
| Preeclampsia | Síndrome de HELLP | Púrpura trombocitopénica | SHUa | |
|---|---|---|---|---|
| Hipertensión | Muy común | Muy común | Raro/ausente | Común |
| Proteinuria | Constante | Constante | Raro/ausente | Constante |
| Ictericia | Raro/ausente | Raro/ausente | Raro/ausente | Raro/ausente |
| Síntomas neurológicos | Común | Muy común | Común | Raro/ausente |
| Dolor abdominal | Raro/ausente | Muy común | Raro/ausente | Raro/ausente |
| Trombocitopenia | Común | Constante | Constante | Constante |
| Hemolisis | Raro/ausente | Constante | Constante | Constante |
| Elevación de la bilirrubina total | Común | Constante | Constante | Constante |
| Elevación de las transaminasas | Común | Constante | Raro/ausente | Raro/ausente |
| en el trimestre | 3° | 3° | 2° o 3° | Puerperio |
SHUa: síndrome hemolítico urémico atípico.
La presencia de una falla renal aguda que amerita manejo con tratamiento sustitutivo renal se encuentra asociada a mutaciones germinales de los genes de FCH, CD 46, FCI30.
El diagnóstico diferencial de las microangiopatías incluye: la coagulación intravascular diseminada, preeclampsia-eclampsia y síndrome de HELLP31.
DiagnósticoEn las pacientes con sospecha clínica de síndrome hemolítico urémico atípico, el primer paso es confirmar la presencia de anemia hemolítica, trombocitopenia y realizar el diagnóstico diferencial con enfermedades que puedan tener estos hallazgos. Los estudios de laboratorio iniciales deben incluir una biometría hemática para documentar la anemia y la trombocitopenia, la presencia de esquistocitos en frotis de sangre, para determinar la presencia de una deshidrogenasa láctica elevada como parte del estudio de anemia hemolítica, la elevación de la creatinina, la determinación de ADAMTS 13, además de un coprocultivo y PCR para E. coli 015732.
El diagnóstico es difícil ya que la mayoría de las pruebas no se encuentran disponibles en los servicios de ginecología31. En pacientes de alto riesgo o con carga genética para el síndrome hemolítico urémico atípico hay que realizar test de ADN buscando mutaciones en C3, FB, FH, FI, CD46. También se proponen pruebas de formulación en diacilglicerol cinasa ¿ (DGK¿), que codifica una proteína que no está en el sistema del complemento32. La determinación de estas pruebas se debe realizar antes de ofrecer un tratamiento transfusional a estas pacientes.
Las pacientes con síndrome hemolítico urémico atípico mediada por complemento presentan niveles bajos de C3 o C4, sin embargo, los niveles plasmáticos dentro de rango de C3, C4, FCB, FCH y TPI no excluyen el diagnóstico de síndrome hemolítico urémico atípico mediado por el complemento33.
TratamientoLa terapia con plasmaféresis o infusión de plasma fresco congelado ha sido la piedra angular de la terapia del síndrome hemolítico urémico atípico desde 1980 y fue esencialmente, la única terapia disponible hasta hace poco; la decisión entre una u otra forma de terapia dependen del tamaño de la paciente y del grado de insuficiencia renal, los cuales limitan la utilización de la transfusión3. Mediante la infusión de plasma fresco congelado se infundían proteínas reguladoras de la vía alternativa del complemento normofuncionantes, mientras que con los recambios plasmáticos se buscaba eliminar: las proteínas disfuncionantes, los anticuerpos anti-FCH y posibles disparadores de la agresión endotelial (factores trombogénicos o inflamatorios)4. En el embarazo el tratamiento inicial es la plasmaféresis, realizada de la misma forma que en las pacientes no embarazadas34, pero antes de comenzar la terapia se recomienda realizar un panel viral para excluir cualquier infección antes de la transfusión e incluso se recomienda la vacunación en pacientes susceptibles35. Se desconoce el esquema terapéutico más eficaz, aunque las guías de consenso recomiendan un inicio precoz, mantenido e intenso, con sesiones diarias de plasmaféresis usando 2 volúmenes plasmáticos en adultos, y con una disminución lenta cuando la cuenta plaquetaria sea mayor de 150×109/l por al menos 3 días, y las concentraciones de deshidrogenasa láctica sean normales. Si no se observa mejoría en un lapso de 5 días, debe considerarse iniciar eculizumab; si existe mejoría, el intervalo debe irse alargando a una semana o cada 2 semanas35. Si no se puede iniciar la plasmaféresis dentro de las primeras 24 h de presentación, se iniciará transfusión de plasma fresco a dosis de 10-20ml/kg si la paciente no muestra datos de sobrecarga de volumen o datos de insuficiencia cardiaca y debe limitarse la dosis en hipertensión y en falla renal. La duración de la terapia y los intervalos entre la dosificación dependerán de la evolución clínica de cada paciente. En algunos artículos, en pacientes embarazadas, se continúa el tratamiento hasta llevar el embarazo a término35. Aunque el parto, por lo general, no causa resolución de púrpura trombocitopénica trombótica y síndrome hemolítico urémico, hay evidencia anecdótica de que puede hacerlo en algunas pacientes cuando cursan con preeclampsia sobreagregada36. Si existe respuesta al tratamiento, el embarazo debe continuarse a término con monitorización obstétrica y considerar la interrupción del embarazo en caso de que se presenten las alteraciones numeradas en la tabla 235–40.
Indicaciones para interrupción del embarazo en el síndrome hemolítico urémico atípico
| Anormalidades hematológicas |
| Trombocitopenia o microangiopatía hemolítica que progresa a pesar del tratamiento |
| Anormalidades neurológicas |
| Estatus mental anormal (confusión, estupor, coma, desorientación) |
| Anormalidades focales (afasia, disartria, déficits motores focales) |
| Anormalidades renales |
| Falla renal aguda |
| Anormalidades fetales |
| Datos de distrés o sufrimiento fetal |
| Perfil biofísico alterado |
| Datos de restricción del crecimiento intrauterino |
En las pacientes con síndrome hemolítico urémico atípico que presentan una mala respuesta al tratamiento se debe considerar el tratamiento con eculizumab. Su mecanismo de acción es por inhibición de la vía del complemento, específicamente inhibiendo al C5 en vía del complemento terminal.
El uso de eculizumab en síndrome hemolítico urémico atípico gestacional es anecdótico. En los últimos 10 años se han publicado 7 artículos de su uso, sin embargo, no existe evidencia suficiente para hacer una recomendación como primera línea de tratamiento. La dosis reportada en la literatura es de 900 mg semanales con intervalos de 2 a 4 semanas, sin que se pueda explicar la causa de esta diferencia entre las dosis, y se continúa con una dosis de mantenimiento de 1,200 mg cada 7-14 días. Todos reportaron remisión completa del síndrome, sin recurrencia a 6 meses de seguimiento41-45. En pacientes con síndrome hemolítico urémico atípico y tratamiento sustitutivo renal prolongado, la recuperación de la función renal se logró con el uso de eculizumab. Existen reportes de remisión clínica de la enfermedad que permitieron la resolución del embarazo en la semana 38 sin repercusiones fetales ni maternas.
PronósticoEste síndrome presenta una elevada morbimortalidad, sin embargo, el diagnóstico temprano y la adecuada monitorización y tratamiento oportuno mejoran el pronóstico. Las pacientes en las cuales se han documentado alteraciones en C3 deben recibir consejería genética debido al riesgo de pérdida fetal y al de desarrollo de preeclampsia. Sin embargo, la identificación de esta mutación no permite predecir el riesgo de manifestar síndrome hemolítico urémico atípico gestacional, debido a que la penetrancia genética de esta enfermedad es variable. Los datos publicados reportan una penetrancia global del 50 al 60% y factores adicionales, como alteraciones en FH y PCM, son los disparadores extras para el desarrollo de síndrome hemolítico urémico atípico. Por lo tanto, cualquier paciente con alteración del complemento deberá ser monitorizada estrictamente por un obstetra con experiencia en embarazo de alto riesgo. No se debe realizar plasmaféresis profiláctica durante el embarazo, aun con la presencia de antecedente de síndrome hemolítico urémico atípico, ya que el riesgo de las complicaciones excede los beneficios de un tratamiento empírico.
ConclusionesLa progresión y el entendimiento de la fisiopatología del síndrome hemolítico urémico atípico en la última década ha logrado obtener nuevas opciones de tratamiento, las cuales puedan ofrecer la prevención de su evolución en pacientes con alto riesgo. Al ser el embarazo un disparador de este síndrome, consideramos necesaria la evaluación de las pacientes de alto riesgo, lo que hace necesario conocer la verdadera prevalencia de esta enfermedad durante la gestación. Se necesitan estudios de las 2 líneas de tratamiento actuales (plasmaféresis y eculizumab) para definir las indicaciones de ambas en pacientes con este síndrome y para conocer su efectividad en la reversión de la enfermedad como monoterapia o en terapia combinada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.