




Gastrointestinal stromal tumors (GIST) are a group of mesenchymal neoplasms that affect the gastrointestinal tract, defined by the expression of the CD117 (c-kit) onco-protein.1 Their appearance outside of the gastrointestinal tract is uncommon.
We present a case of primary hepatic extra-gastrointestinal stromal tumor (EGIST) in an adult patient with a negative extension study for another primary tumor, who was treated surgically, progressed favorably and is currently disease free.
The patient is a 41-year-old male with no personal history of interest. He was under study for abdominal discomfort and weight loss in previous months. Upon examination, a mass was observed in the hypochondrium and right flank, which reached the right anterior superior iliac spine. Complete blood count was normal, with the following biochemistry results: cholesterol=222mg/dL (HDL=69mg/dL); total bilirubin=1.65g/dL at the expense of indirect (1.38mg/dL) and GGT=102U/L. Liver enzymes, coagulation and tumor markers (α-fetoprotein, CEA, CA-125, CA 15.3 and CA 19.9) were normal.
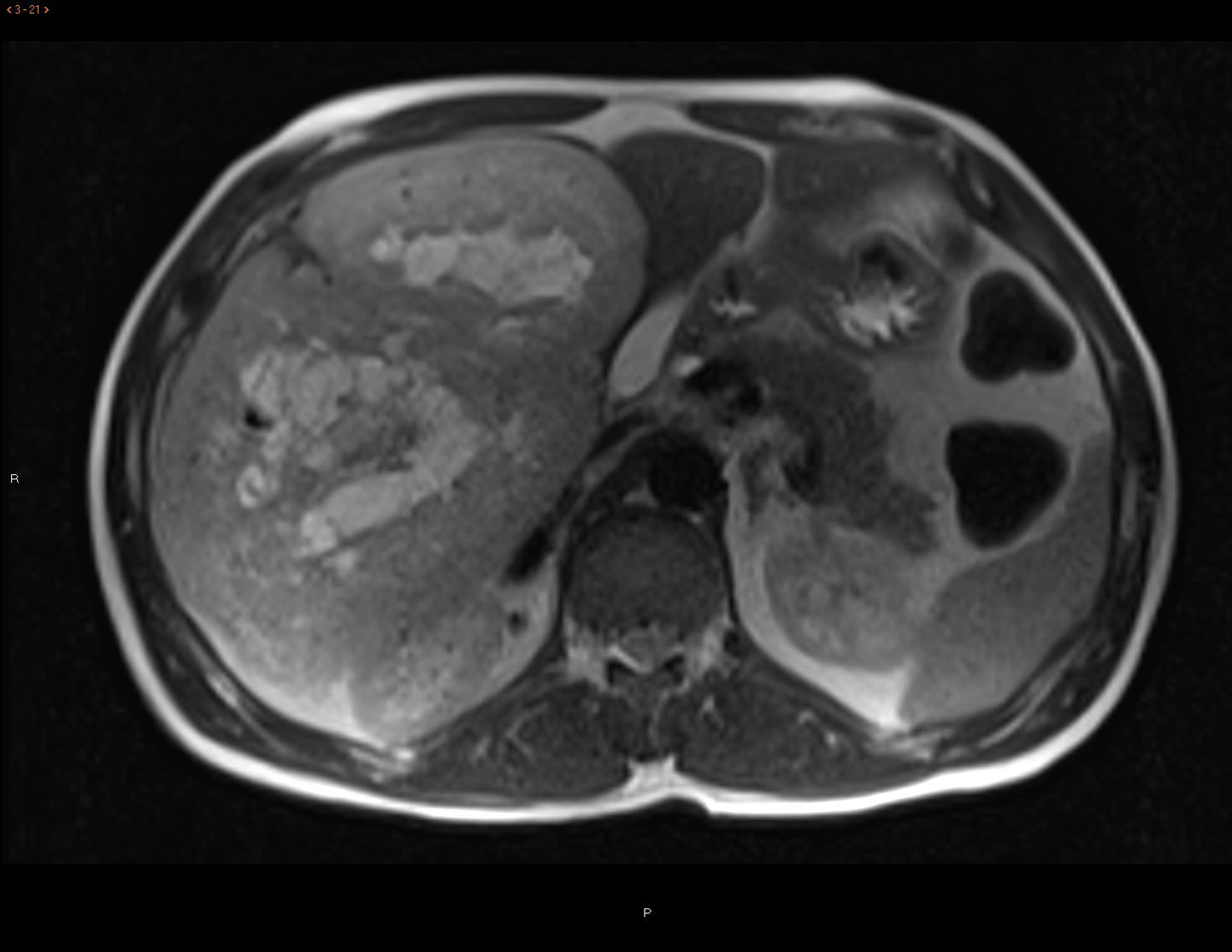
CT scan and MRI showed a mass measuring 20×19.5×13.6cm in length with significant contrast uptake, central necrotic areas and exophytic location (liver segments V and VI), with no infiltration of neighboring organs. The differential diagnosis included hypervascular tumors (adenoma, hypervascular metastasis, sarcoma) (Fig. 1). Gastroscopy and colonoscopy were normal. PET/CT showed no extrahepatic uptake.

The patient underwent surgery, which revealed a hypervascularized encapsulated tumor with terminal involvement of the right suprahepatic branch. A segmentectomy was performed of liver segments V-VI, with an intraoperative blood loss of 600cc due to this vascularization, requiring a single Pringle maneuver of 23min.
The postoperative period transpired with type A liver failure, and the patient required the transfusion of 2 units of packed red blood cells. The patient was discharged from hospital on the 8th day post-op, with no further incidents.
The definitive histological diagnosis was: grade 1 GIST tumor, with spindle cells that infiltrated and destroyed the liver parenchyma, leaving a few nests of hepatocytes in the middle of the tumor growth (pT4). The mitotic rate was 5 mitoses per 50 high-power fields and the proliferative index (Ki-67) was 10%, with free surgical margins. The differential diagnosis included leiomyosarcoma and synovial sarcoma, with positivity for DOG-1, CD117 and Bcl-2, while desmin, S100 and CD34 were negative. The mutation analysis demonstrated deletion of exon 9 of the c-kit gene, confirming the diagnosis of GIST (Fig. 2).
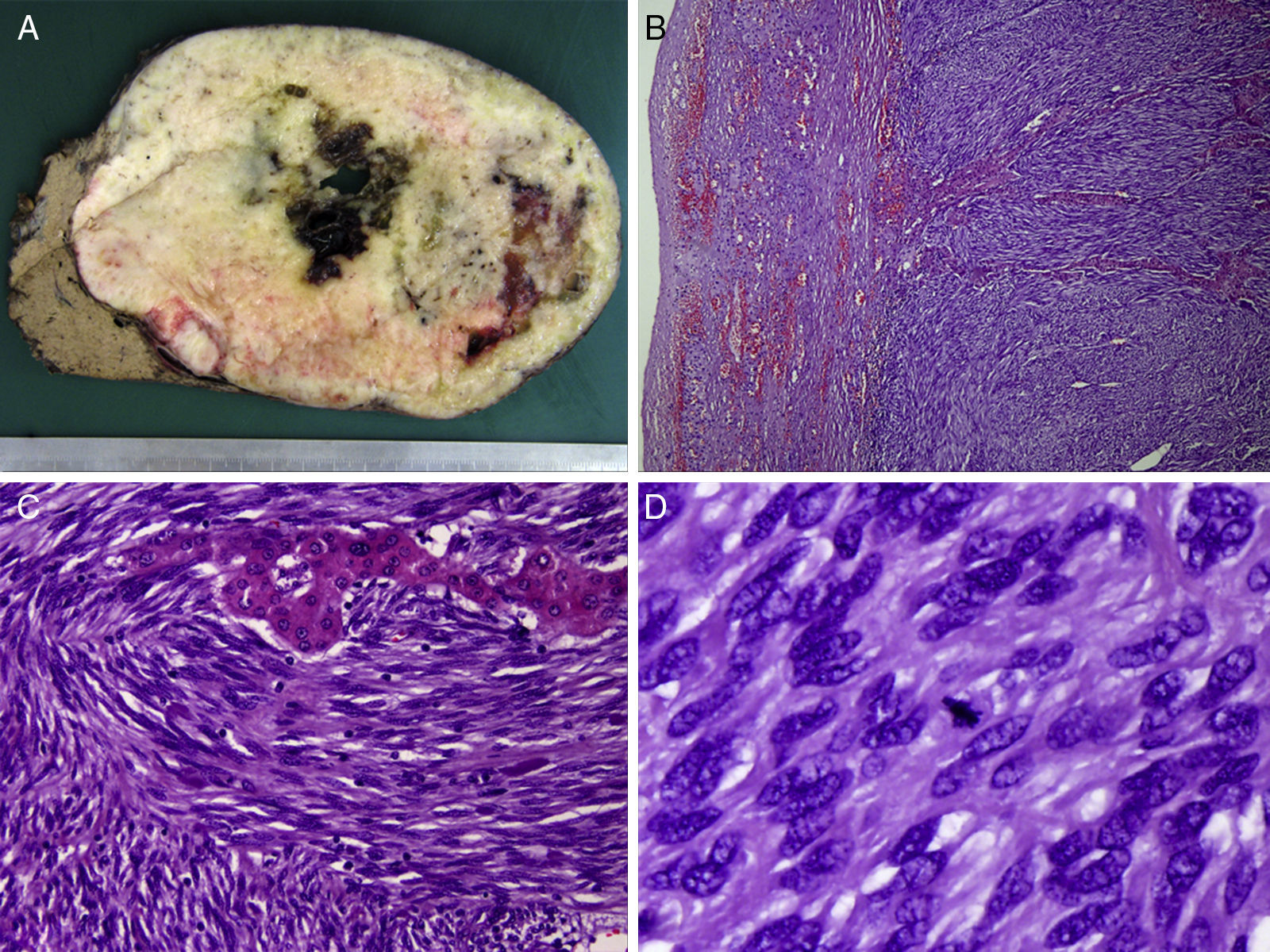
 Macroscopic image of the specimen, with a firm whitish mass with well-defined edges, a fleshy cut surface and foci of cystic degeneration, measuring 19cm in diameter; (B) microscopic image (hematoxylin-eosin ×100) of spindle cells arranged in bundles, with eosinophil cytoplasm and elongated nucleus surrounding a trabecula of hepatocytes; (C) strong and diffuse cytoplasmic expression of c-kit (CD117), characteristic in this type of tumors; and (D) positive staining for Bcl-2 expression.")
(A) Macroscopic image of the specimen, with a firm whitish mass with well-defined edges, a fleshy cut surface and foci of cystic degeneration, measuring 19cm in diameter; (B) microscopic image (hematoxylin-eosin ×100) of spindle cells arranged in bundles, with eosinophil cytoplasm and elongated nucleus surrounding a trabecula of hepatocytes; (C) strong and diffuse cytoplasmic expression of c-kit (CD117), characteristic in this type of tumors; and (D) positive staining for Bcl-2 expression.
After surgery, the patient initiated adjuvant therapy with imatinib at higher doses than usual due to the presence of risk factors (size, mutation exon 9, Ki-67), and the follow-up PET/CT showed no evidence of hypermetabolic lesions suggestive of recurrence. Currently, the patient continues to be asymptomatic and in complete remission after 18 months of follow-up.
Presently, GIST tumors are defined as fusiform or epithelioid CD117-positive mesenchymal tumors that are primary tumors of the gastrointestinal tract, mesentery and retroperitoneum. GIST were first described in 1983 by Mazur and Clark to designate non-epithelial tumors of the digestive tract lacking smooth muscle ultrastructural features and immunohistochemical characteristics of Schwann cells.1 GIST are the most common sarcoma of the gastrointestinal tract, accounting for 2% of gastrointestinal tumors, but 80% of sarcomas of this origin. The stomach is the most frequent location. Its incidence is 10 to 20 cases per million inhabitants/year.2–4 The maximum incidence is in the fourth and sixth decades; the distribution by gender is similar; although some studies suggest a predominance among males.2,3 GIST share immunophenotypic similarities with interstitial cells of Cajal and characteristics such as expression of CD117 glycoprotein, CD34, smooth muscle myosin heavy chain and nestin5 are common, although there is controversy about the origin from pluripotent stem cells.
The differential diagnosis includes morphologically similar or CD34-positive lesions (fibrohistiocytic tumors, peripheral nerve sheath tumors, Kaposi's sarcoma, etc.) and tumors that express c-kit (melanoma, dedifferentiated liposarcoma, undifferentiated small cell lung cancer).4,5
The presence of metastasis and/or invasion of neighboring organs are criteria for malignancy. As for the molecular biology, the prognostic importance of c-kit mutations in exons 11, 9, 13 and 175–7 is being assessed, along with other factors such as VEGF expression, loss of CD44 expression, p16 alterations and genetic markers (genetic overexpression of VIL2, COL8A1, CCNB2, HMG2, TSG101, etc.).7
Regarding tumor aggressiveness, all authors agree on the influence of tumor size and the mitotic index, establishing prognostic groups.8,9 The determination of cell proliferation with Ki-67 staining is also used (index of 10% or more is associated with a poor prognosis).
GIST that appear outside of the gastrointestinal tract are very uncommon and are typically found in one single organ. Cases have been reported in the pancreas, liver, omentum, prostate, seminal vesicle, pleura, etc., although they are rare. These are classified as EGIST.
The primary location in the liver is exceptional, and there are no protocols to assess the risk for malignancy in this location. In our case, gastroscopy and colonoscopy were normal, CT and PET/CT showed no evidence of extrahepatic disease, and there was no evidence of recurrence after 18 months of follow-up. Therefore, we concluded that the disease was a primary hepatic EGIST, whose size would classify it as “high” risk if it had been located elsewhere.9,10
The authors would like to thank all the Management Units at the Complejo Hospitalario in Jaén (Spain) involved either directly or indirectly in the management of the present clinical case, especially Dr. Dabán Collado of the General and Digestive Surgery Departments. Likewise, they would like to thank the members of the Hepatobiliary-Pancreatic Tumor Committee at the Complejo Hospitalario in Jaén.
Please cite this article as: Carrillo Colmenero AM, Serradilla Martín M, Redondo Olmedilla MD, Ramos Pleguezuelos FM, López Leiva P. Tumor del estroma extragastrointestinal primario hepático gigante. Cir Esp. 2017;95:547–550.
