




Despite current standards of care, a considerable risk of atherosclerotic cardiovascular disease remains in both primary and secondary prevention. In this setting, clonal hematopoiesis driven by somatic mutations has recently emerged as a relatively common, potent and independent risk factor for atherosclerotic cardiovascular disease and other cardiovascular conditions. Experimental studies in mice suggest that mutations in TET2 and JAK2, which are among the most common in clonal hematopoiesis, increase inflammation and are causally connected to accelerated atherosclerosis development, which may explain the link between clonal hematopoiesis and increased cardiovascular risk. In this review, we provide an overview of our current understanding of this emerging cardiovascular risk factor.
A pesar de los estándares de los cuidados actuales, persiste un riesgo considerable de enfermedad cardiovascular aterosclerótica tanto en la prevención primaria como secundaria. En este contexto, la hematopoyesis clonal impulsada por mutaciones somáticas ha emergido recientemente como un factor de riesgo relativamente común, potente e independiente, para la enfermedad cardiovascular aterosclerótica y otras situaciones cardiovasculares. Los estudios experimentales en ratones sugieren que las mutaciones en TET2 y JAK2, que se encuentran dentro de las hematopoyesis clonales más comunes, incrementan la inflamación y están causalmente conectadas con el desarrollo de aterosclerosis acelerado, lo cual puede explicar el vínculo entre la hematopoyesis clonal y el incremento del riesgo cardiovascular. En esta revisión, aportamos una visión general de nuestra comprensión actual de este factor de riesgo cardiovascular.
The exposure over the years to traditional cardiovascular risk factors, particularly hypercholesterolemia, is undeniably the main driver of atherosclerotic cardiovascular disease. Yet, imaging studies show that atherosclerosis can progress in individuals who are at low risk based on conventional prediction algorithms.1–3 Additionally, clinical and epidemiological evidence demonstrate that a significant risk of atherosclerotic cardiovascular disease (CVD) remains even when traditional cardiovascular risk factors seem managed properly.4,5 The mechanisms underlying this so-called residual risk are currently the objective of intensive research, as they hold promise for the development of new strategies to improve cardiovascular risk management. In this setting, the clonal expansion of hematopoietic cells that bear certain acquired mutations is emerging as an important new contributor to atherosclerotic CVD.
Somatic mutations in the hematopoietic system and clonal hematopoiesisMutations can be classified as either those arising in germ cells, which are inherited by the progeny (i.e. germline mutations), or those acquired during life by non-germ cells (i.e. somatic mutations). Human genetic studies during the last two decades have unveiled a contribution of many inherited variants to atherosclerotic CVD. Now, some somatic mutations are also emerging as potent contributors to CVD.
With advances in tissue sampling techniques and high-throughput DNA sequencing technologies, it is increasingly recognized that carrying somatic mutations is not the exception, but the normal, for the vast majority of human tissues.6,7 These mutations are typically linked to the origin of cancer, but their pathophysiological implications have become a topic of increasing interest beyond the oncology field. In this context, the hematopoietic system has been studied to the greatest extent, partly thanks to the easy access to peripheral blood samples and the availability of extensive blood sequencing datasets from large cohorts. Based on estimates of mutation rates8 and the typical number of hematopoietic stem cells (HSC) in humans, a middle-aged individual may carry on the order of 1 million mutations in the HSC pool. This sets the stage for a competition among the different mutant clones, which leads to the selection of mutations that provide a fitness advantage to HSCs. In this context, whereas most mutations are neutral or deleterious to HSC function, some mutations provide an advantage by promoting self-renewal, proliferation or survival of the mutant HSC, which leads to the progressive expansion of such mutant clones. This phenomenon can be described as somatic mutation-driven clonal hematopoiesis. Importantly, this clonal expansion initially occurs in the HSC population within the bone marrow, but it progressively has a reflection in its progeny (immune cells, red blood cells, and platelets), whose dysfunction plays central roles in a variety of diseases.
Clonal hematopoiesis is typically identified through next-generation DNA sequencing of blood samples, which allows for the detection of clonally expanded mutations based on the calculation of variant allele fractions (VAF, see key terminology in Table 1). The catalog of mutations that can be detected in blood is vast, including base substitutions (single-nucleotide variants, SNVs), small insertions and deletions (indels), cytogenetic aneuploidies and structural chromosomal variants.9–15 Accordingly, clonal hematopoiesis can be defined in different manners based on the type of mutations and several technical parameters (Table 1). However, the definition that is gaining popularity, particularly in the cardiovascular field, is that of clonal hematopoiesis of indeterminate potential or CHIP. In the literature, CHIP is defined as the presence in blood or bone marrow of an expanded SNV or indel in a known hematological malignancy-related gene at a VAF of at least 2%, without meeting the criteria for diagnosis of hematological disease.16 CHIP mutations can be acquired randomly at any point in life, even soon after the formation of the zygote, but this phenomenon is strongly associated with aging because the chances of having acquired such mutations evidently increase as an individual ages and their expansion is expected to be slow and require years. CHIP has been estimated to be present in 2–3% of middle-aged individuals and in 10–20% of those older than age 70.10,13 However, these numbers probably represent an underestimation of the prevalence of CHIP, as they are mainly based on analyses of whole exome/genome sequencing datasets, which provide limited sensitivity for the detection of CHIP mutations, as further discussed below.
Key terms related to clonal hematopoiesis.
| Clonal hematopoiesis | Condition whereby a substantial fraction of an individual's blood cells is derived from a single dominant hematopoietic stem cell clone, in contrast to normal hematopoiesis, which is typically polyclonal. Clonal hematopoiesis can be driven by acquired mutations, but also by nonmutational mechanisms.11,41,42 Overt hematologic malignancies may also be included in this definition, as they are also the result of a clonally restricted hematopoietic process. |
| Somatic mutation-driven clonal hematopoiesis | Clonal hematopoiesis driven by que acquisition of a mutation that provides a competitive advantage to the mutant hematopoietic stem cell, leading to its clonal expansion. We use this term to draw a distinction with non-mutational drivers of clonality in the hematopoietic system.42 |
| Age-related clonal hematopoiesis (ARCH) | Clonal expansion of hematopoietic stem cells carrying recurrent genetic variants in individuals without clear diagnosis of hematological malignancies. This term does not include any specific criteria for variant allelic fraction or the presence of a putative leukemia-driver mutation.43 |
| Clonal hematopoiesis of indeterminate potential (CHIP) | Presence of somatic mutations in myeloid cancer-associated genes in the blood or bone marrow of individuals without a blood cancer or other known hematological abnormality. In CHIP, the mutant clone must be detected with a variant allele fraction greater than 2%, although this threshold might be revised. This term was proposed as an entity to distinguish preleukemic clonal hematopoiesis from hematological malignancies and ‘benign’ forms of clonal hematopoiesis, either malign or benign.16 |
| Variant Allele Fraction (VAF) | Percentage of reads that support a mutant allele out of the total number of reads in a next-generation sequencing study. It is frequently used as an estimate of mutant clone size; assuming a mutation is monoallelic, a VAF of 10% means that ∼20% of the cells from a given sample contain the mutation. |
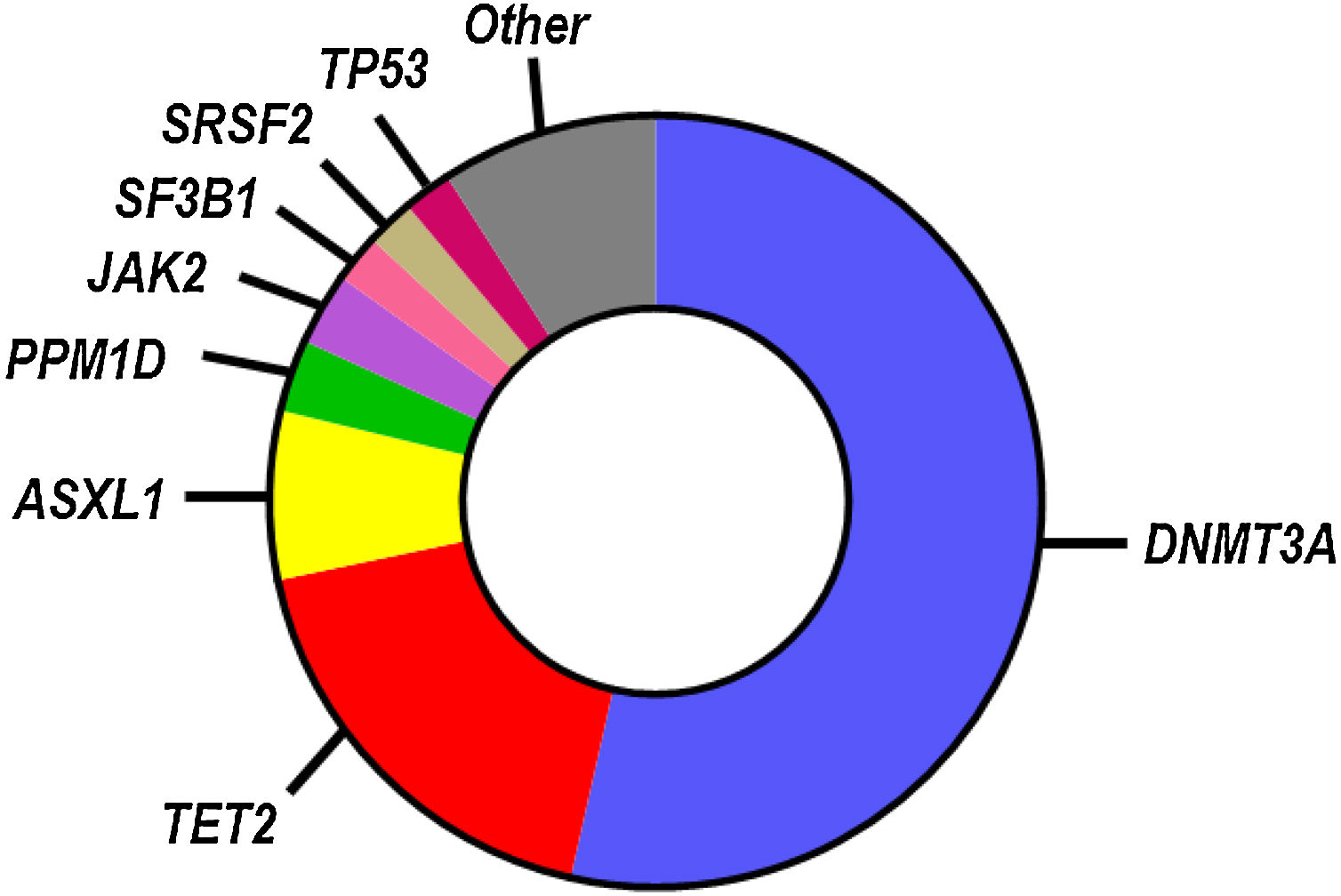
While SNVs and indels in >100 blood cancer-related genes have been identified as candidate drivers of clonal hematopoiesis, most known CHIP mutations occur in a small subset of genes (Fig. 1), most frequently in those encoding the epigenetic regulators DNMT3A, TET2 and ASXL1.9–13 Other frequently mutated genes linked to CHIP encode for DNA damage response proteins (TP53, PPM1D), splicing factors (SF3B1, SRSF2) and signaling mediators (JAK2). Individuals who exhibit CHIP typically carry only one, or, less frequently, two or three mutations, and they have a substantially increased relative risk of developing hematological disease.9,10 However, the absolute risk of developing hematological malignancies remains modest even in CHIP-mutation carriers, ranging from 0.5% to 1% per year, which probably reflects the need to acquire multiple cooperative oncogenic mutations for the malignant transformation of the mutant clone. Hence, most individuals who exhibit CHIP never develop hematologic malignancies and exhibit normal blood cell counts. CHIP is, however, associated with increased all-cause mortality, due to its strong connection with atherosclerotic CVD and other cardiovascular conditions (Fig. 2), which has led to the recognition of this phenomenon as a new cardiovascular risk factor.

Mutational spectrum in CHIP. The most commonly mutated genes in CHIP based on whole genome sequencing analysis are shown in the chart.13 The relative number of mutations in each gene is proportional to its representation. This mutational spectrum may vary, if CHIP is investigated in specific age ranges or by more sensitive sequencing approaches.

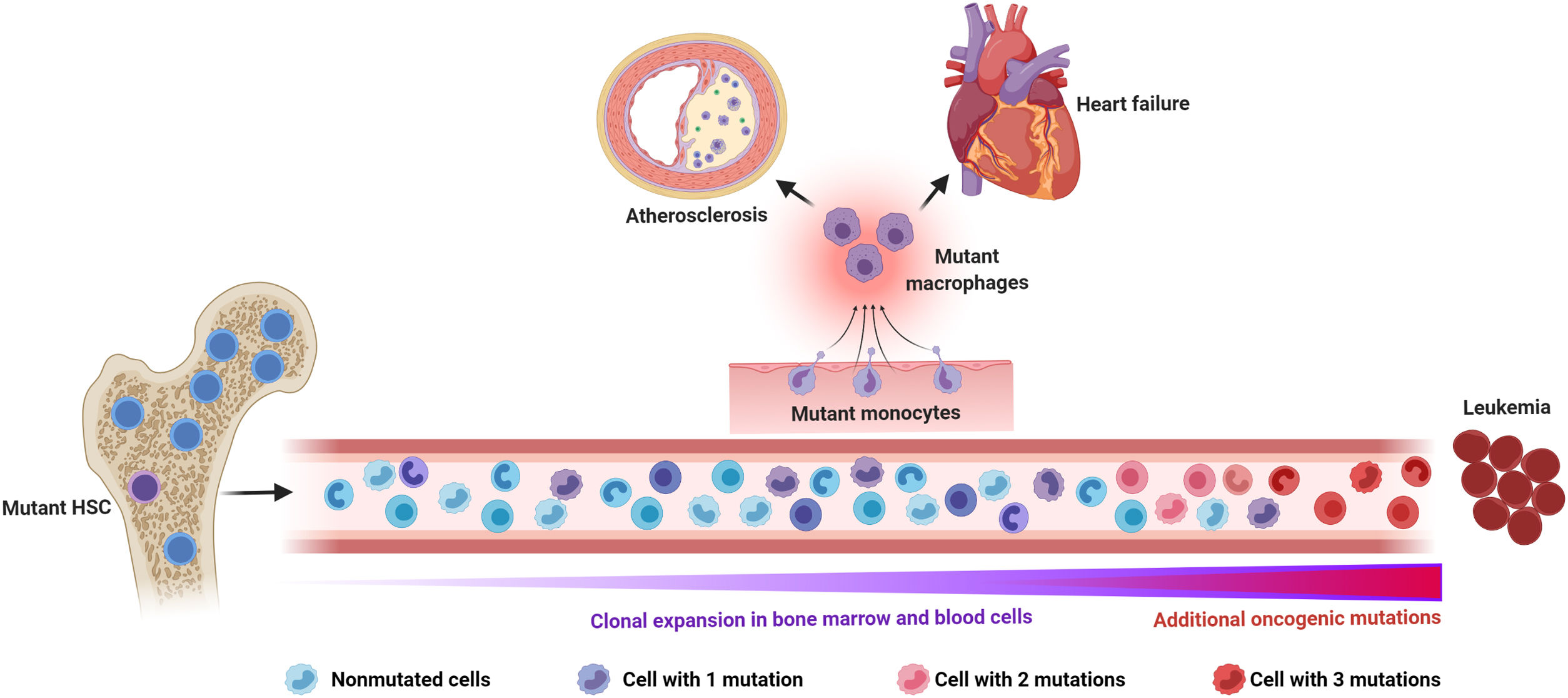
Somatic mutations and clonal hematopoiesis: shared risk factors for hematological cancer and cardiovascular disease. The random acquisition and accumulation of somatic mutations in hematopoietic stem cells is an inevitable consequence of normal aging. Some of these mutations confer a competitive advantage to the mutant cell, leading to clonal hematopoiesis, which in most cases is driven by one single mutation. While this situation increases the risk of developing a hematologic malignancy, this typically requires the acquisition of multiple mutations, which is infrequent, even in individuals with clonal hematopoiesis. The main cause of death in individuals exhibiting clonal hematopoiesis is cardiovascular disease due to the association of this phenomenon, at least when driven by certain mutations, with atherosclerotic cardiovascular disease and adverse clinical progression of heart failure. Increased inflammatory responses by mutant monocytes/macrophages are emerging as key contributors to the elevated cardiovascular risk in clonal hematopoiesis, although additional cell types may also play an important role in this context. Figure created with BioRender.com.
The initial finding of an association between CHIP and atherosclerotic CVD came from an unplanned secondary analysis of whole exome sequencing datasets in a study intended to detect pre-leukemic mutations.10 An exploratory analysis of these data revealed that CHIP was associated with elevated all-cause mortality mainly due to CVD. Further analyses led to the unexpected finding that those with CHIP exhibited a >2-fold increased risk of developing coronary heart disease and ischemic stroke, even after adjustment for known CVD risk factors.10 The association between CHIP and atherosclerotic CVD was subsequently replicated in case–control cohorts for coronary heart disease,17 as well as in other population cohorts.18,19 Recent studies have also unveiled a robust association between CHIP and higher risk of incident heart failure20 and adverse clinical outcomes in patients with ischemic or non-ischemic heart failure with reduced left ventricular ejection fraction (Fig. 2).21–23
While CHIP is typically identified through a meta-analysis of mutations in a compendium of candidate clonal hematopoiesis driver genes, a single gene analysis suggested an association between increased coronary heart disease risk and somatic mutations in DNMT3A, TET2, ASXL1 and JAK2,17 which will need to be replicated in independent studies. Whether mutations in other frequently mutated genes, such as TP53 or SF3B1, are individually associated with atherosclerotic CVD risk remains unknown. Yet, importantly, while mutations in different genes may have distinct impact on CVD, the magnitude of risk conferred by CHIP overall has been reported to be as great as or even greater than that of many conventional risk factors for CVD.10,17 Hence, CHIP mutations have the potential to be potent CVD risk modifiers.
Clonal hematopoiesis as a driver of atherosclerosisThe human genetic association between CHIP and atherosclerotic CVD has opened up exciting discussions on the possibility of developing new strategies for CVD risk management. In this context, it is biologically plausible that mutated blood cells are related causally to CVD because the most common of these mutations occur in genes that play broad roles in regulating essential cellular functions. However, the inherently descriptive nature of genetic association studies demands that these findings are interpreted cautiously, as they do not allow to determine whether CHIP and CVD are causally linked or whether CHIP is simply a marker of aging or other confounding phenomena. While much work lies ahead, laboratory studies and observations in humans have started to shed light onto causality in the relationship between CHIP and CVD.
An increasing burden of evidence strongly supports the possibility that CHIP, at least when driven by certain mutations, accelerates the development of atherosclerosis. In humans, in an analysis of a small subset of participants in the BioImage Study, individuals with CHIP mutations exhibited greater coronary artery calcium scores, a radiological surrogate of atherosclerosis burden.17 In mice, the availability of strains that exhibit genetic alterations in the murine orthologues of the most frequently mutated human genes is enabling the testing of causality in experimental atherosclerosis studies. To date, two CHIP genes have been investigated using this approach, Tet2 and Jak2, and available evidence strongly support a causal contribution of somatic mutations in these genes to atherosclerosis.
Tet2, which encodes for an epigenetic regulator of gene transcription,24 was the first gene reported to exhibit somatic mutations in blood cells in individuals with clonal hematopoiesis without blood cancer,25 and loss-of-function mutations in this gene are among the most common in CHIP.9–11,13 Using competitive bone marrow transplantation studies in atherosclerosis-prone Ldlr−/− mice, we demonstrated that either biallelic (−/−) or monoallelic (+/−) inactivation of Tet2 lead to accelerated atherosclerosis development, in the absence of quantitative differences in blood cell counts.26 Analyses of the effects of pan-hematopoietic TET2 ablation in an independent study reached highly concordant findings,17 and additional animal studies suggest a similar contribution of TET2-mutant cells to insulin resistance27 and heart failure.28,29 Mechanistic studies with hematopoietic- and myeloid-specific TET2-deficient mice and primary macrophages suggest that accelerated atherosclerosis in conditions of TET2 loss of function mainly results from the pro-inflammatory activity of TET2-mutant macrophages,17,26 characterized predominantly by an upregulation of IL-1β production at several levels. TET2 inactivation markedly increases IL-1β transcript levels in macrophages exposed to a variety of pro-inflammatory stimuli, through a mechanism mediated, at least in part, by increased histone acetylation at the Il1b promoter.26 Furthermore, TET2-deficient macrophages exhibit increased activity of the NLRP3 inflammasome,26 a main mediator of IL-1β maturation and secretion.30 Accordingly, TET2-deficient macrophages exhibit a remarkable increase in IL-1β secretion,26,27 which exceeds the elevation of its transcript levels, and pharmacological NLRP3 inhibition suppresses the effects of TET2-mutant cells on experimental atherosclerosis26 and other conditions.27,28 Consistent with these findings, human studies show that circulating IL-1β levels are significantly elevated in TET2 mutation carriers.13 Such elevation is not observed in carriers of other CHIP mutations, suggesting a specific effect of somatic TET2 mutations on the production of this pro-inflammatory and pro-atherogenic cytokine.
In contrast to TET2 mutations, the JAK2V617F hotspot variant linked to CHIP is a gain-of-function mutation, which results in constitutive activation of the JAK2 signaling kinase and downstream mediators. This mutation is strongly associated with myeloproliferative neoplasms such as polycythemia vera and essential thrombocytosis, but can also be detected in individuals with no apparent hematological abnormalities, in whom it associated with a disproportionate risk of atherosclerotic cardiovascular disease,17 despite correlating with lower circulating cholesterol levels.31 Pan-hematopoietic JAK2V617F expression in Ldlr−/− mice accelerates atherosclerosis, in parallel with a complex hematological phenotype that includes expansion of hematopoietic stem and progenitor cells, leukocytosis, erythrocytosis, thrombocytosis and neutrophilia.32 Because this is essentially a myeloproliferative neoplasm phenotype, more refined models with lineage-specific expression of this mutation have been developed to simulate the human scenario of benign clonal hematopoiesis. In a recent study, Cre/LoxP strategies and S100A8-Cre and CX3CR1-Cre mouse strains were employed to achieve neutrophil- and monocyte/macrophage-specific JAK2V617F expression, respectively.33 This approach revealed that expression of this mutant protein in monocyte/macrophages, but not in neutrophils, increases atherosclerotic plaque size, as well as necrotic core extension within the plaque. Mechanistically, this accelerated atherosclerosis was linked to increased expression of the double-stranded DNA-sensing inflammasome AIM2. Genetic ablation of AIM2, but not that of NLRP3, reduced the effects of JAK2V617F expression on plaque size and necrotic cores, suggesting that activation of the AIM2 inflammasome is a key pathway promoting atherosclerosis in conditions of JAK2V617F-mutant clonal hematopoiesis. Consistent with this possibility and the known role of AIM2 in mediating production of the IL-18 cytokine,34,35 humans bearing the JAK2V617F have been reported to exhibit higher circulating levels of IL-18.13
The differences in atherosclerosis phenotypes and underlying molecular mechanisms observed in mouse models of TET2-mutant and JAK2-mutant clonal hematopoiesis support the idea that mutations in different genes are not equivalent, and that the clinical significance of CHIP most likely depends on the specific mutated gene. Future research will be required to address whether other CHIP mutations, beyond those affecting TET2 and JAK2, are causally linked to accelerated atherosclerosis development.
Clonal hematopoiesis in cardiovascular risk management: Next stepsThe robust human genetic association between CHIP and atherosclerotic CVD, together with the solid data coming from murine models, has opened up exciting discussions on the possibility of using CHIP as the basis to develop new strategies for atherosclerotic CVD risk management,36 particularly as it is a risk factor shared with hematological malignancies and, potentially, several other age-related conditions. Indeed, a number of institutions have established specialized clinics for counseling patients with clonal hematopoiesis.37 However, CHIP screening is not yet recommended in the context of CVD, as there is an insufficient evidence base to inform the management of cardiovascular risk in CHIP carriers. A number of key important questions will need to be addressed to establish guidelines for clinical management of CHIP, which are summarized next.
First, the threshold of mutant clone size that associates with increased atherosclerotic CVD risk remains unclear. The majority of human genetic evidence linking CHIP to cardiovascular risk are based on the analysis of whole exome/genome datasets.10,17–19 However, this strategy provides limited sensitivity to detect somatic mutations. CHIP is typically defined with a VAF threshold of 2%,16 but the analysis of whole exome/genome sequencing data misses a substantial number of mutations with VAF between 2% and 10% (i.e. 4–20% mutant blood cells).13 Taking into consideration this limitation, there is sufficient evidence base to conclude that CHIP with VAF>10% is associated with elevated atherosclerotic CVD risk. However, whether smaller mutant clones, which are much more common, are sufficient to confer an increased risk remains uncertain. Further sequencing efforts with more sensitive approaches will be required to fill this important gap in knowledge.
Second, the directionality of the relationship between CHIP and atherosclerotic CVD is still a matter of debate. While mouse and human studies strongly support a direct contribution of some CHIP mutations to atherosclerosis development, recent mathematical modeling of clonal hematopoiesis dynamics suggests that atherosclerosis can accelerate clonal hematopoiesis, to the extent that reverse causality could explain the CHIP/CVD association according to some investigators.38 Considering all available evidence, the scenario that emerges is a pernicious cycle in which atherosclerosis facilitates clonal hematopoiesis, which, in turn, accelerates the progression of atherosclerosis and the transition to ischemic events. However, this possibility remains speculative.39 Determining whether causality, reverse causality or bi-directionality underlies the CHIP/atherosclerosis connection will be crucial to develop strategies to manage CVD risk in carriers of CHIP mutations. New experiments in mice and longitudinal sequencing studies in human cohorts might help to answer this important question.
Third, we lack evidence-based interventions to prevent the heightened cardiovascular risk associated with CHIP. There is still insufficient information to assess whether the standard of care in atherosclerotic CVD (e.g. lifestyle modifications, cholesterol lowering drugs) prevents the increased cardiovascular risk in CHIP mutation carriers. In this context, targeting the inflammatory pathways hyperactivated in mouse models of clonal hematopoiesis is an attractive possibility, which is already being evaluated through post hoc analyses of completed trials with anti-inflammatory drugs.40 Yet, ultimately, new clinical trials will be required to test the value of personalized preventive care strategies tailored to the effects of specific CHIP mutations. The design of such trials will be challenging, considering the heterogeneity of mutations and VAFs in CHIP, but it will be crucial if we intend to translate our knowledge of this emerging CVD risk factor into improvements in risk management.
ConclusionsSomatic mutations that drive clonal hematopoiesis are emerging as a potent cardiovascular risk factor, which is common in the elderly and may contribute to residual cardiovascular risk. However, our understanding of the link between clonal hematopoiesis and atherosclerosis is incomplete. While evidence to date suggest that heightened CVD risk in individuals who harbor these mutated clones is frequently related to increased inflammation, there is a great need for further investigation into the specific effects of the various mutations linked to clonal hematopoiesis. Both basic and clinical research efforts will be required to develop strategies for the management of this newly recognized contributor to atherosclerotic CVD.
FundingJJF is supported by a Ramón y Cajal award (RYC-2016-20026) from the Spanish Ministerio de Ciencia e Innovación (MICIN)/Agencia Estatal de Investigación (AEI)/10.13039/501100011033 and Fondo Social Europeo “El FSE invierte en tu futuro”. The CNIC is supported by the MICIN, the Instituto de Salud Carlos III, the Pro-CNIC Foundation, and is a Severo Ochoa Center of Excellence (grant CEX2020-001041-S funded by MICIN/AEI/10.13039/501100011033).
Conflict of interestThe authors declare that they have no conflict of interest.