




Vascular smooth muscle cells (VSMCs) constitute the principal cellular component of the medial layer of arteries and are responsible for vessel contraction and relaxation in response to blood flow. Alterations in VSMCs can hinder vascular system function, leading to vascular stiffness, calcification and atherosclerosis, which in turn may result in life-threatening complications. Pathological changes in VSMCs typically correlate with chronological age; however, there are certain conditions and diseases, such as Hutchinson-Gilford progeria syndrome (HGPS), that can accelerate this process, resulting in premature vascular aging. HGPS is a rare genetic disorder characterized by severe VSMC loss, accelerated atherosclerosis and death from myocardial infarction or stroke during the adolescence. Because experiments with mouse models have demonstrated that alterations in VSMCs are responsible for early atherosclerosis in HGPS, studies on this disease can provide insights into the mechanisms of vascular aging and assess the relative contribution of VSMCs to this process.
Las células del músculo liso vascular (CMLV) constituyen el principal componente celular de la capa medial arterial, siendo responsables de la contracción y relajación de los vasos en respuesta al flujo sanguíneo. Las alteraciones en CMLV dificultan la función vascular, generan rigidez vascular, calcificación y aterosclerosis, pudiendo resultar en complicaciones mortales. Cambios patológicos en CMLV suelen correlacionarse con la edad cronológica; sin embargo, existen afecciones y enfermedades, como el síndrome de progeria de Hutchinson-Gilford (HGPS), que pueden acelerar este proceso, provocando envejecimiento vascular prematuro. El HGPS es un trastorno genético raro caracterizado por una pérdida grave de CMLV, aterosclerosis acelerada y muerte por infarto de miocardio o ictus durante la adolescencia. Experimentos con modelos de ratón demostraron que las alteraciones en CMLV son responsables de la aterosclerosis temprana en HGPS. Por tanto, estudios sobre esta enfermedad pueden proporcionar información sobre los mecanismos del envejecimiento vascular y caracterizar la contribución de las CMLV.
Cardiovascular disease (CVD) is a major contributor to mortality worldwide and its incidence and prevalence correlate with increasing age.1,2 Pathological changes in the arteries typically precede clinical manifestations of CVD, and thus have been extensively studied to improve prevention and treatment of CVD. In particular, vascular smooth muscle cells (VSMCs), which are the main cellular component of the artery wall, have received much attention because of their key role in maintaining biomechanical properties of blood vessels. Alterations in VSMCs, due to aging or in response to an injury, promote vascular stiffening, calcification and atherosclerosis, which may lead to myocardial infarction or stroke.3
Vascular disease onset, progression and manifestation vary among individuals.4 Both genetic and environmental factors can modulate this process. One of the most striking examples of accelerated vascular aging is Hutchinson-Gilford progeria syndrome (HGPS).5 HGPS patients develop vascular calcification, stiffening and atherosclerosis during childhood and typically die from its complications before reaching adulthood. Human and mouse model data indicate that most vascular pathologies in progeria stem from VSMC defects, suggesting that HGPS is a valuable model to study VSMC aging.6 In this review, we will focus on the role of VSMCs in vascular disease associated with HGPS. We will summarize the main findings in humans and mouse models, outline molecular and cellular mechanisms underlying VSMC alterations and discuss treatment strategies to delay premature vascular aging in progeria.
Molecular mechanism underlying HGPSHGPS is an extremely rare disease caused by mutations in the LMNA gene.7,8 In healthy differentiated cells, the LMNA gene gives rise to two major isoforms, lamin A and lamin C, via alternative splicing. Lamin C is encoded by exons 1-10 of LMNA gene and does not undergo post-translational modifications. Lamin A is encoded by all 12 exons of LMNA and is produced as a precursor protein, called prelamin A. To reach its mature form, the C-terminus of prelamin A is farnesylated by farnesyl transferase, then its last three amino acids are cleaved off. Next, the newly exposed cysteine residue is carboxymethylated by isoprenyl carboxyl methyltransferase (ICMT). The final step in lamin A maturation is the cleavage of 15 residues containing farnesyl and carboxymethyl modifications by ZMPSTE24 metalloprotease. Lamin A is then incorporated in the nuclear lamina, where it is crucial for maintaining proper structure and function of the nucleus.
Most HGPS cases are caused by a de novo point mutation (c.1824C>T; p.G608G) in exon 11 of LMNA that only affects the lamin A isoform.7,8 Although this single nucleotide substitution does not change the encoded amino acid, it activates a cryptic splice site leading to production of a shorter mRNA. The resulting lack of 150 nucleotides in the LMNA mRNA removes a 50-amino-acid region in the prelamin A protein, including the cleavage site for ZMPSTE24. Hence, the mutant protein, called progerin, cannot undergo complete post-translational processing. Unlike lamin A, progerin remains permanently farnesylated and carboxymethylated and stays anchored to the inner nuclear membrane, causing nuclear defects. Abnormal nuclear structure and function lead to cellular and tissue malfunction that ultimately manifests as organismal aging.
Vascular disease in HGPS patientsHGPS patients present many signs that resemble physiological aging, including baldness, loss of subcutaneous fat, scleroderma, joint rigidity and osteolysis.9 However, the most relevant symptoms of the disease are linked to the cardiovascular system, as they provoke death at 14.6 years of age on average.5,10 Children with HGPS showed an increase in both carotid-femoral pulse wave velocity and arterial wall ecodensity, indicating vascular stiffening.11 In some patients, elevated blood pressure was observed.11,12 Postmortem examination of HGPS arteries revealed a series of alterations, such as VSMC depletion in the media, increased extracellular matrix (ECM) deposition, elastin fragmentation and adventitial thickening.5,13,14 Moreover, calcification was found in the arterial media, plaques and valves of HGPS subjects.5,13 Importantly, autopsies of affected individuals revealed advanced, often acellular and fibrotic, atherosclerotic lesions with signs of disruption.5,13 Accordingly, death in about 90% of HGPS cases occurred from atherosclerosis complications, mainly ischemic heart disease.9 Even though cerebrovascular accidents account for only 10% of HGPS-related deaths,15 60% of patients showed evidence of strokes, out of which half were clinically silent.16 Remarkably, most HGPS individuals did not show classical CVD risk factors, such as increased levels of low-density lipoprotein (LDL) or C-reactive protein.17 In summary, HGPS features CVD similar to that observed during normal aging and might constitute a valuable model to study mechanisms of premature vascular decline.
Mouse models to study VSMC aging in progeriaGiven the low number of patients living with progeria worldwide and limited availability of postmortem tissue samples, several animal models of HGPS have been generated to study disease mechanisms and search for a treatment. The first HGPS animal model was created by introducing a bacterial artificial chromosome (BAC) containing the human LMNA gene carrying the G608G mutation into a wild-type mouse with two normal Lmna alleles.18 Although hemizygote BAC-G608G mice did not show overall aging phenotype, they presented vascular abnormalities starting at 5 months of age that included VSMC loss, elastic fiber breakage, proteoglycan accumulation, collagen deposition and adventitial thickening. At 16 months of age, BAC-G608G mice also showed medial calcification. When bred to homozygosity, BAC-G608G mice presented reduced lifespan and a more severe phenotype that also included symptoms outside the cardiovascular system.19
Subsequent models were generated by introducing an HGPS-equivalent mutation in the mouse Lmna gene (c.1827C>T; p.G609G) in order to produce progerin via aberrant splicing. These models, all referred to as LmnaG609G, displayed general aging features, such as lipodystrophy, reduced bone density, failure to thrive and shortened survival.20–22 Both LmnaG609G/+ and LmnaG609G/G609G mice showed similar symptoms, but disease onset was earlier and progression faster in homozygotes.20,22,23 Aorta of the LmnaG609G/G609G mice showed a mild reduction in VSMC content, increased collagen deposition, altered elastin waving, moderate adventitial thickening and arterial stiffening.20,23–25 Although incipient medial calcification was observed in LmnaG609G/G609G mice, it fully manifested in the heterozygous mice probably due to their longer survival.26,27
However, none of the abovementioned models presented atherosclerotic disease, the death-causing progeria symptom. This observation is in agreement with the fact that mice, unlike humans, are resistant to atherogenesis because of their atheroprotective lipid profile.28 To enable atherogenesis, LmnaG609G/G609G mice were crossed with Apoe- or Ldlr-deficient mice, two widely-used models to study atherosclerosis. Both Apoe−/−LmnaG609G/G609G and Ldlr−/−LmnaG609G/G609G mice presented accelerated atherosclerotic disease along with severe VSMC loss and collagen deposition in the media, and excessive adventitial thickening.6,29 Furthermore, aortas of Apoe−/−LmnaG609G/G609G mice showed lipid accumulation in the medial layer that was, at least partially, attributed to increased LDL retention.6 Importantly, atheroma plaques of Apoe−/−LmnaG609G/G609G and Ldlr−/−LmnaG609G/G609G mice showed signs of vulnerability, including reduced VSMC content in the plaque and in the fibrous cap and presence of erythrocytes and iron deposits.6,29 Likewise, evidence of myocardial infarction was found in a few Apoe−/−LmnaG609G/G609G mice close to their maximum survival.6 In summary, both atheroprone ubiquitous HGPS models aged prematurely and closely recapitulated the cardiovascular phenotype found in human patients.
To further disentangle the role of VSMCs in progerin-induced vascular aging, tissue-specific models of HGPS were generated both with and without atherogenic background. Namely, LmnaLCS/LCSSM22αCre mice expressing progerin predominantly in VSMCs exhibited alterations in the aortic media, including VSMC loss, collagen accumulation and straightened elastin layers.6,24 Likewise, these mice showed reduced vessel contractility and increased vascular stiffening.24,30 However, this model did not show atherosclerosis nor shortened lifespan.6 When LmnaLCS/LCSSM22αCre mice were crossed to Apoe-deficient background, they manifested the complete vascular phenotype with premature atherosclerotic disease, similar to that observed in Apoe−/−LmnaG609G/G609G mice.6,31 At advanced stages of the disease, Apoe−/−LmnaLCS/LCSSM22αCre mice also showed excessive plaque calcification, a feature not found in other HGPS mouse models. Notably, Apoe−/−LmnaLCS/LCSSM22αCre mice, despite not showing general premature aging features, had a reduced survival. Given the evidence of myocardial infarction found in these mice close to their maximum survival, this early mortality was probably due to atherosclerosis complications. These findings demonstrated that VSMC alterations underlie the accelerated vascular aging in HGPS.
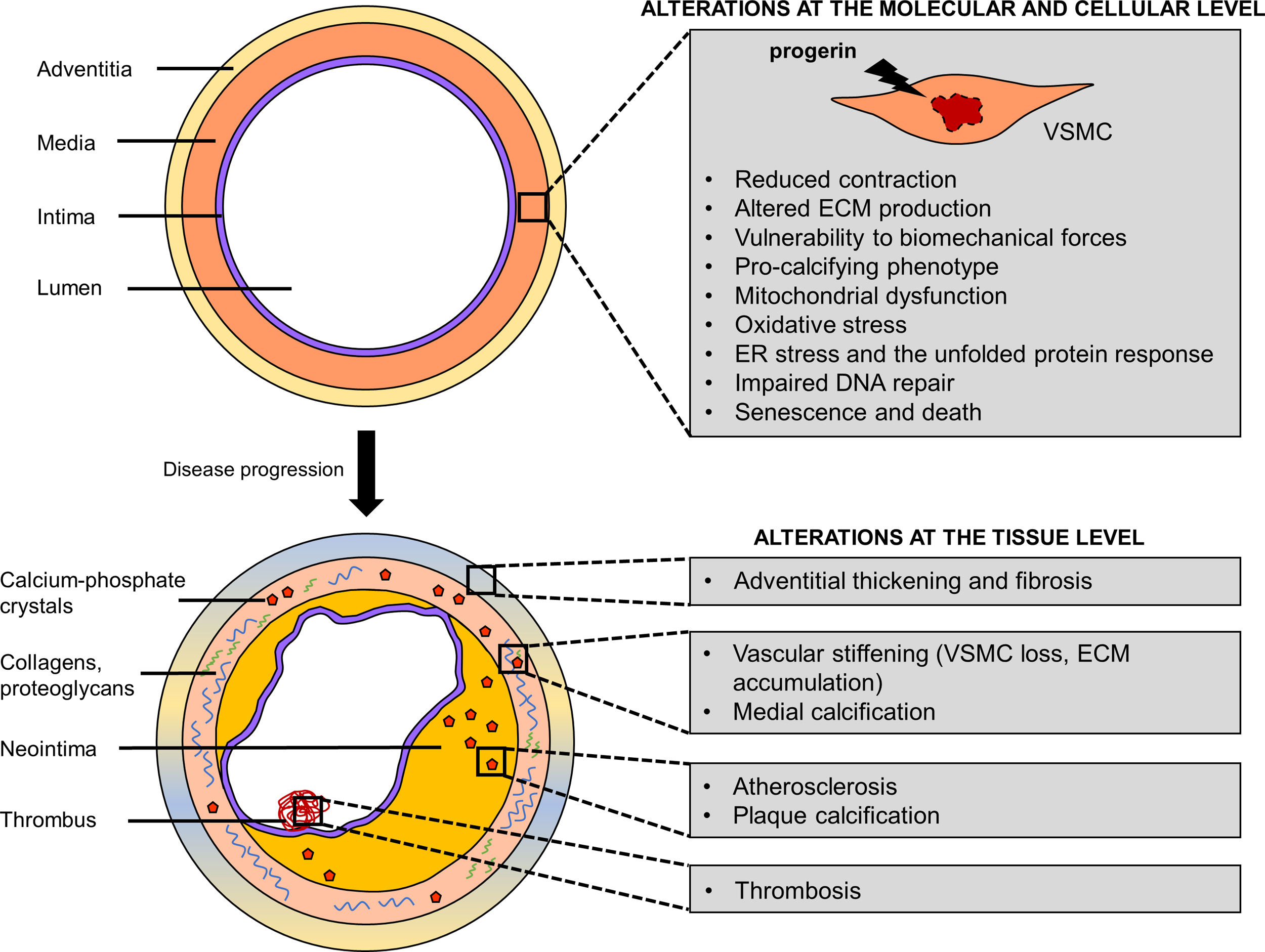
Cellular and molecular mechanisms of progerin-induced VSMC agingProgerin induces multiple cellular and molecular defects in VSMCs that with time translate into vascular pathologies, including stiffness, calcification and atherosclerosis (Fig. 1). VSMCs are among the cell types that are most severely affected by progerin expression. This sensitivity could be partially explained by the fact that lamin A (and thus progerin) levels are greatly increased in stiff tissues, such as aorta.23,32
 defects underlie premature vascular disease in Hutchinson-Gilford progeria syndrome. Progerin expression in VSMCs triggers a series of molecular and cellular defects (impaired contraction, altered extracellular matrix production/remodeling, vulnerability to biomechanical forces, osteo/chondrogenic phenotype, defective DNA repair, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress and the unfolded protein response) that lead to VSMC senescence and death. Changes in VSMC function and their loss in the arteries result in vascular stiffness, calcification, adventitial thickening, atherosclerosis and its life-threatening complications, such as thrombosis. ECM, extracellular matrix; ER, endoplasmic reticulum.")
Vascular smooth muscle cell (VSMC) defects underlie premature vascular disease in Hutchinson-Gilford progeria syndrome. Progerin expression in VSMCs triggers a series of molecular and cellular defects (impaired contraction, altered extracellular matrix production/remodeling, vulnerability to biomechanical forces, osteo/chondrogenic phenotype, defective DNA repair, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress and the unfolded protein response) that lead to VSMC senescence and death. Changes in VSMC function and their loss in the arteries result in vascular stiffness, calcification, adventitial thickening, atherosclerosis and its life-threatening complications, such as thrombosis. ECM, extracellular matrix; ER, endoplasmic reticulum.
VSMCs are subjected to repetitive biomechanical stress related to blood flow. In mouse models of progeria, aortic zones with disturbed blood flow and dynamic changes in shear stress typically suffer more pronounced vascular disease. For instance, the inner curvature of LmnaG609G/G609G and LmnaG609G/+ mouse aortas showed more severe VSMC depletion than the outer curvature despite expressing similar levels of progerin.23In vitro studies showed that progerin-expressing VSMCs were more susceptible to cell death when subjected to repetitive biaxial stretch. Recently, it was reported that vulnerability to biomechanical forces of VSMCs derived from HGPS induced pluripotent stem cells (iPSCs) is mediated by an upregulation of matrix metalloprotease-13.33
VSMCs regulate vascular tone by contracting and relaxing in response to blood flow. Ex vivo analysis of LmnaG609G/G609G and LmnaLCS/LCSSM22αCre aortas by wire myography showed increased stiffening and reduced contractility.24,30 Transcriptomic analysis of descending aortas of young LmnaG609G/G609G mice revealed changes in several pathways associated with contractility.34 Furthermore, LmnaG609G/G609G aortas showed reduced mRNA levels of Myh11, the gene encoding smooth muscle-specific myosin heavy chain (SM-MHC), one of the main proteins responsible for the contractile properties of VSMCs. Immunofluorescence analysis of carotid arteries from LmnaG609G/G609G mice confirmed lower levels of SM-MHC in the medial layer.
VSMCs also play a vital role in maintaining and remodeling the ECM of blood vessels. Medial aortas of progeroid mice at advanced disease stages showed quantitative and qualitative changes in ECM composition, including increased collagen deposition, altered elastin layers and proteoglycan accumulation.18,23,24,29 Transcriptomic analysis of medial aortas from Apoe−/−LmnaG609G/G609G and Apoe−/−LmnaLCS/LCSSM22αCre mice before the onset of the disease revealed profound alterations in the expression of genes related to fibrosis.35 Similarly, upregulation of collagen III and lysyl oxidase, a collagen crosslinking enzyme, was found in the medial layer of aorta and carotid arteries of young LmnaG609G/G609G mice, coinciding with vascular stiffening.25
VSMCs modulate vascular calcification by secreting either inhibitors of calcium-phosphate crystal deposition in normal conditions or pro-osteo/chondrogenic molecules upon pathological phenotypic switching. Some progeria models presented medial or intimal calcification, possibly attributable to both VSMC loss (and hence loss of calcification inhibitors) and phenotypic changes in this cell type.6,26,27 Calcified aortas from LmnaG609G/+ mice were found to present augmented mRNA expression of osteogenic markers Bmp2 and Runx2.26 Moreover, VSMCs derived from LmnaG609G/+ mice showed reduced capacity to inhibit calcification in vitro due to reduced levels of extracellular pyrophosphate resulting from both its augmented degradation and diminished production. Increased degradation of pyrophosphate was associated with upregulation of tissue-nonspecific alkaline phosphatase (TNAP), an enzyme hydrolyzing pyrophosphate. Decreased pyrophosphate production was linked to impaired production of ATP, which is the main substrate for pyrophosphate synthesis, and increased activity of ectonucleoside triphosphate diphosphohydrolase-1 (eNTPD1), an enzyme degrading ATP to phosphate. Studies with cultured VSMCs from LmnaG609G/+ mice revealed that reduced ATP production resulted from increased reactive oxygen species (ROS) production and mitochondrial dysfunction.36
VSMCs, together with endothelial cells and macrophages, are key players in atherogenesis. VSMCs typically migrate toward the growing neointima, then proliferate and synthetize ECM to form a fibrous cap that protects plaques from rupture. In atheroprone models of progeria, VSMC loss in the media correlated with reduced VSMC content in the plaque and in the fibrous cap and with plaque disruption.6,29 RNA sequencing of medial aortas from Apoe−/−LmnaG609G/G609G and Apoe−/−LmnaLCS/LCSSM22αCre mice prior to vascular disease manifestation revealed changes in pathways related to NRF2-mediated oxidative stress, endoplasmic reticulum (ER) stress and the unfolded protein response.35 Immunofluorescence analyses of the medial layer of both aforementioned models confirmed augmented expression of ER stress markers, such as calreticulin, protein disulfide isomerase and binding-immunoglobulin protein. Further experiments suggested that unresolved ER stress and the associated unfolded protein response are likely to trigger VSMC death in the arteries, accelerating atherosclerosis in HGPS.
Finally, VSMC senescence and death might carry serious consequences for blood vessel structure and function. Almost all progeria models showed progressive loss of VSMCs in the arteries. Consistent with this, p16, a senescence marker, could already be detected in the arterial media of young LmnaG609G/G609G mice.25In vitro studies with VSMCs concluded that progerin causes lower proliferation and replication rates, and triggers senescence due to impaired DNA repair.36–38 Moreover, HGPS-iPSC VSMCs in culture showed prolonged mitosis and died by mitotic catastrophe.38 Taken together, these experiments suggest that progerin expression in VSMCs induces a series of interconnected defects leading to phenotype switching and death, which prompt premature vascular aging.
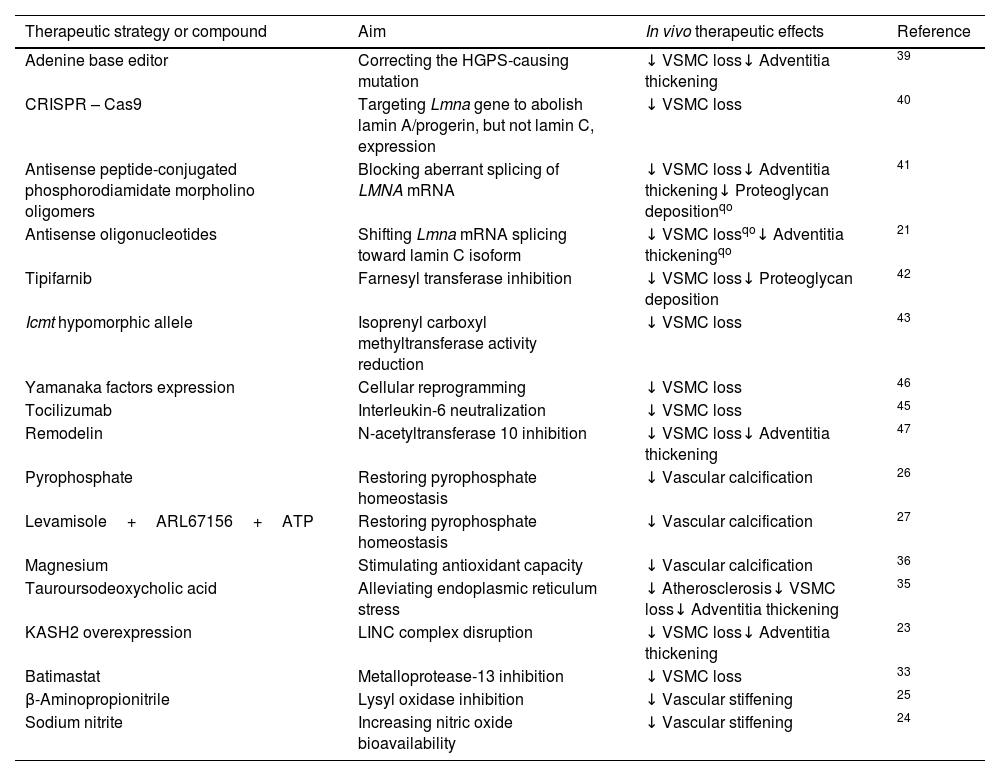
Therapeutic strategies to delay vascular aging in mouse models of HGPSNumerous basic research studies have proposed possible therapeutic approaches to treat progeria, ranging from gene editing strategies to modulators of downstream effects of progerin. However, only a fraction of these studies explored the impact of these treatments on vascular aging in vivo (Table 1).
In vivo therapeutic strategies to counteract vascular disease in mouse models of Hutchinson-Gilford progeria syndrome (HGPS).
| Therapeutic strategy or compound | Aim | In vivo therapeutic effects | Reference |
|---|---|---|---|
| Adenine base editor | Correcting the HGPS-causing mutation | ↓ VSMC loss↓ Adventitia thickening | 39 |
| CRISPR – Cas9 | Targeting Lmna gene to abolish lamin A/progerin, but not lamin C, expression | ↓ VSMC loss | 40 |
| Antisense peptide-conjugated phosphorodiamidate morpholino oligomers | Blocking aberrant splicing of LMNA mRNA | ↓ VSMC loss↓ Adventitia thickening↓ Proteoglycan depositionqo | 41 |
| Antisense oligonucleotides | Shifting Lmna mRNA splicing toward lamin C isoform | ↓ VSMC lossqo↓ Adventitia thickeningqo | 21 |
| Tipifarnib | Farnesyl transferase inhibition | ↓ VSMC loss↓ Proteoglycan deposition | 42 |
| Icmt hypomorphic allele | Isoprenyl carboxyl methyltransferase activity reduction | ↓ VSMC loss | 43 |
| Yamanaka factors expression | Cellular reprogramming | ↓ VSMC loss | 46 |
| Tocilizumab | Interleukin-6 neutralization | ↓ VSMC loss | 45 |
| Remodelin | N-acetyltransferase 10 inhibition | ↓ VSMC loss↓ Adventitia thickening | 47 |
| Pyrophosphate | Restoring pyrophosphate homeostasis | ↓ Vascular calcification | 26 |
| Levamisole+ARL67156+ATP | Restoring pyrophosphate homeostasis | ↓ Vascular calcification | 27 |
| Magnesium | Stimulating antioxidant capacity | ↓ Vascular calcification | 36 |
| Tauroursodeoxycholic acid | Alleviating endoplasmic reticulum stress | ↓ Atherosclerosis↓ VSMC loss↓ Adventitia thickening | 35 |
| KASH2 overexpression | LINC complex disruption | ↓ VSMC loss↓ Adventitia thickening | 23 |
| Batimastat | Metalloprotease-13 inhibition | ↓ VSMC loss | 33 |
| β-Aminopropionitrile | Lysyl oxidase inhibition | ↓ Vascular stiffening | 25 |
| Sodium nitrite | Increasing nitric oxide bioavailability | ↓ Vascular stiffening | 24 |
qo, qualitative observation; LINC, linker of nucleoskeleton and cytoskeleton; VSMC, vascular smooth muscle cell.
Since progerin elicits its toxic effects in a dominant-negative manner, several preclinical therapeutic strategies aimed at reducing progerin expression. The most upstream approach for treating progeria is correction of the HGPS-causing mutation using an adenine base editor (ABE) that converts the aberrant A•T base pair to a G•C base pair, as in the wild-type LMNA allele. ABE treatment in BAC-G608G homozygous mice at P14 completely abolished medial VSMC depletion and adventitial fibrosis, and extended lifespan 2.4 times compared to saline-treated controls, representing the most efficient treatment described to date.39 Another gene-based therapy involved Lmna gene disruption using CRISPR-Cas9 technology to specifically prevent expression of lamin A and progerin, without altering lamin C production. Intravenous administration of guide RNAs to LmnaG609G/G609G-Cas9 neonates alleviated VSMC loss and increased survival by 25%.40 RNA splicing is another therapeutic target for reducing progerin expression. Thus, administration of antisense peptide-conjugated phosphorodiamidate morpholino oligomers, which blocked the LMNA exon 11–12 aberrant splicing, ameliorated the aortic phenotype of BAC-G608G homozygous mice, and extended their lifespan by 61.6%.41 Likewise, an antisense oligonucleotide therapy shifting Lmna splicing toward lamin C (at the expense of lamin A and hence progerin) was able to partially prevent VSMC loss and adventitial thickening in LmnaG609G/G609G mice.21
Given that the retention of post-translational modifications is partially responsible for progerin detrimental properties, several studies explored the possibility of inhibiting farnesylation and carboxymethylation. Administration of tipifarnib, a farnesyl transferase inhibitor, to BAC-G608G hemizygous mice delayed VSMC loss and proteoglycan deposition in the aorta.42 Similarly, reducing ICMT activity by introducing a hypomorphic Icmt allele into LmnaG609G/G609G mice recovered the number of VSMCs in their aorta, and increased their body weight and survival.43
Since lamin A is involved in regulating numerous cellular processes, progerin affects a wide range of biological pathways. Hence, other therapeutic strategies are focused on targeting either systemic or cell/tissue-specific effects of progerin expression. For example, progerin triggers inflammation that manifests as increased interleukin-6 concentrations in serum.44 Consistently, administration of interleukin-6-neutralizing antibodies to LmnaG609G/G609G mice protected from medial VSMC loss and provided a modest lifespan extension.45 Moreover, partial cellular reprogramming by expression of Yamanaka factors in LmnaG609G/G609G mice alleviated VSMC depletion in the aorta and extended survival.46 Likewise, inhibition of N-acetyltransferase 10 (NAT10) by remodelin prolonged lifespan, normalized aortic VSMC content and diminished adventitial thickening of LmnaG609G/G609G mice.47 However, the exact link between progerin and NAT10 has not been established yet.
Other treatments for HGPS aimed at restoring pyrophosphate homeostasis. Namely, systemic injection of exogenous pyrophosphate reduced vascular calcification without affecting body weight or survival of LmnaG609G/G609G mice.26 To increase the levels of pyrophosphate in serum, this therapeutic approach was further improved by combining ATP with levamisole and ARL67156, TNAP and eNTPD inhibitors, respectively.27 This combined treatment blocked vascular calcification and provided modest benefits in terms of weight and survival in LmnaG609G/+ mice.
Some compounds used in preclinical studies targeted the negative effects of cellular stress responses triggered by progerin. In this regard, supplementing drinking water with magnesium prevented vascular calcification and improved body weight and survival in LmnaG609G/+ mice by enhancing antioxidant activity and reducing mitochondrial ROS production.36 Furthermore, intraperitoneal administration of tauroursodeoxycholic acid (TUDCA), a bile acid that alleviates ER stress, was able to delay VSMC loss and adventitial thickening in the aorta of Apoe−/−LmnaG609G/G609G and Apoe−/−LmnaLCS/LCSSM22αCre mice.35 Remarkably, TUDCA was the only compound shown to inhibit atherosclerosis in progeroid mice.
Other strategies specifically focused on ECM, contraction and/or mechanosensing in progerin-expressing VSMCs. Disruption of the linker of nucleoskeleton and cytoskeleton complex by VSMC-specific KASH2 overexpression in LmnaG609G/G609G mice partially rescued VSMCs in the media and reduced adventitial thickening.23 Similarly, in vivo inhibition of matrix metalloprotease-13 by batimastat increased VSMC counts in LmnaG609G/G609G medial aortas.33 Furthermore, administration of β-aminopropionitrile to block lysyl oxidase protected LmnaG609G/G609G mice from vascular stiffening.25 Finally, sodium nitrate in drinking water was able to reduce vascular stiffening in LmnaG609G/G609G mice probably due to increased nitric oxide bioavailability.24
Although multiple potential treatments for HGPS have been proposed by basic research laboratories, only one, lonafarnib (an inhibitor of farnesyl transferase), was approved by the Food and Drug Administration. Given that lonafarnib provides only modest therapeutic benefits,10,48,49 more research is required to reach more effective treatments for HGPS. Until strategies aimed at reducing progerin expression become safe in humans, the most plausible approach to treat HGPS would require a combination of several treatments targeting different downstream effects of progerin.
Are studies on HGPS relevant for physiological vascular aging?HGPS shares many pathological traits with physiological aging, especially those related to the cardiovascular system, such as vascular stiffening, calcification and atherosclerosis.5,11 Like in the general population, the leading cause of death in HGPS patients is myocardial infarction or stroke. Postmortem examination of blood vessels revealed that HGPS atheroma plaques frequently exhibit calcification and signs of plaque rupture or erosion, similarly to lesions in normally aged individuals.5 Even though severe depletion of VSMCs in the media and excessive adventitial thickening are hallmarks of HGPS,5,13,14 slightly reduced medial VSMC content and adventitial thickening near atheroma plaques have been reported during normal aging.50,51 Interestingly, low levels of progerin were detected in cells and tissues, including arteries, from old non-HGPS individuals, suggesting that progerin might play a role during normal aging.5
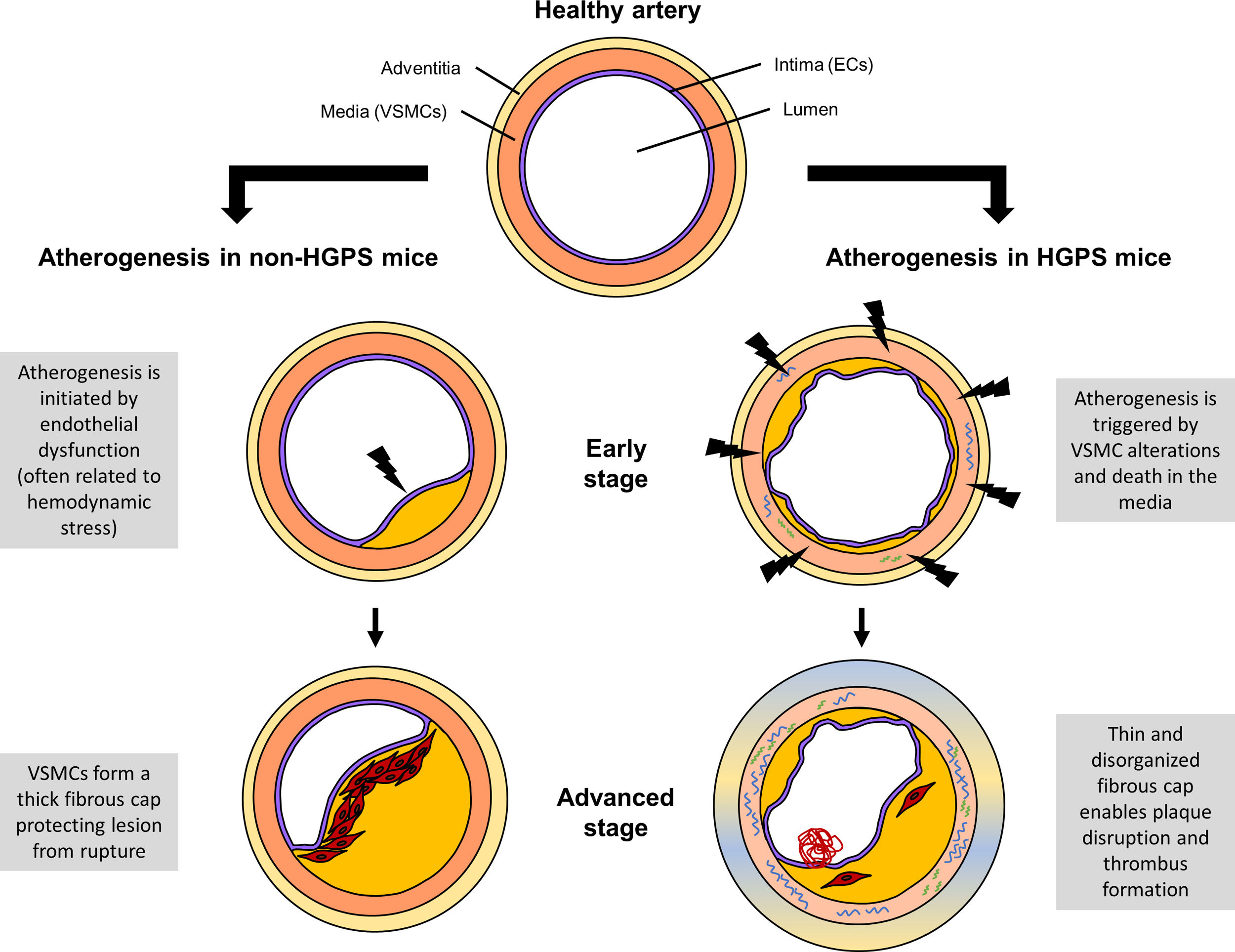
On the contrary, HGPS patients do not suffer several characteristics of aging, such as enhanced cancer susceptibility or dementia.15 In terms of vascular disease, HGPS seems to differ from physiological aging by the principal stimulus that triggers atherogenesis. Namely, mouse model data indicate that in HGPS animals atherosclerosis originates from VSMC alterations and death whereas in non-HGPS subjects it is typically initiated by endothelial dysfunction (Fig. 2).6,29 As a consequence, atheroma lesions in progeria mice form throughout the entire arterial surface while in non-HGPS animals plaques are usually found at specific locations, such as branching sites, predisposed to atherogenesis by disturbed blood flow. Furthermore, HGPS patients and progerin-expressing mice do not show elevated LDL levels in blood, a key CVD risk factor in the general population.6,17,29 Yet, increased retention of LDL in the aortic wall has been found in Apoe−/−LmnaG609G/G609G and Apoe−/−LmnaLCS/LCSSM22αCre mice, suggesting that despite normal systemic levels of LDL, their local concentration in the artery wall is increased and contributes to early atherosclerosis.6 In summary, progerin-driven vascular disease shares many features with age-related arteriosclerosis; however, extrapolation of some results might be limited by the existence of progeria-specific mechanisms.
 and non-HGPS mouse models. In non-HGPS animals (Apoe−/− and Ldlr−/−), atherosclerosis typically begins with endothelial dysfunction, often associated with hemodynamic stress. Activated endothelial cells (ECs) recruit immune cells to the artery wall, leading to atheroma lesion growth. Vascular smooth muscle cells (VSMCs) start to migrate from the media toward the intima, where they proliferate and synthesize extracellular matrix to form a fibrous cap preventing plaque disruption. In HGPS animals (Apoe−/−LmnaG609G/G609G and Ldlr−/−LmnaG609G/G609G), atherogenesis is triggered by alterations in VSMCs and their death in the medial layer. The consequent VSMC depletion in the fibrous cap leads to highly unstable atheroma lesions that can easily break, resulting in life-threatening thrombus formation.")
Comparison of atherogenesis in Hutchinson-Gilford progeria syndrome (HGPS) and non-HGPS mouse models. In non-HGPS animals (Apoe−/− and Ldlr−/−), atherosclerosis typically begins with endothelial dysfunction, often associated with hemodynamic stress. Activated endothelial cells (ECs) recruit immune cells to the artery wall, leading to atheroma lesion growth. Vascular smooth muscle cells (VSMCs) start to migrate from the media toward the intima, where they proliferate and synthesize extracellular matrix to form a fibrous cap preventing plaque disruption. In HGPS animals (Apoe−/−LmnaG609G/G609G and Ldlr−/−LmnaG609G/G609G), atherogenesis is triggered by alterations in VSMCs and their death in the medial layer. The consequent VSMC depletion in the fibrous cap leads to highly unstable atheroma lesions that can easily break, resulting in life-threatening thrombus formation.
Due to a remarkable extension of the average life expectancy, the number of aged individuals is rapidly growing worldwide,52 which in turn prompts a variety of socio-economic and healthcare challenges. These problems predominantly originate from the increased incidence and prevalence of age-associated diseases, such as cancer and CVD. Despite the progress in prevention and treatment, CVD continues to be the principal cause of death in most countries, indicating that our understanding of the mechanisms governing cardiovascular aging is insufficient.1,2
Rare genetic diseases featuring premature CVD, especially those that manifest during childhood, offer a unique opportunity to study mechanisms of accelerated vascular aging isolated from lifestyle factors. In HGPS, one of the most severe premature aging syndromes, atherosclerotic complications represent the main cause of early mortality.5 Premature vascular decline in HGPS is mainly attributed to VSMC alterations, as VSMC-specific expression of progerin in Apoe−/− mice is sufficient to trigger the complete manifestation of progeria-associated vascular phenotype.6,31 VSMC aging induced by progerin involves decreased contractility, altered ECM production, impaired mechanosensing, pro-calcifying phenotype and activation of several cellular stress responses, which in turn lead to cell senescence and death. Altered phenotypes of VSMCs and their loss in arteries result in vascular stiffening, calcification and atherosclerosis, which ultimately contribute to mortality. Similar alterations at the cellular and tissue levels have been described during normal aging,3 suggesting that HGPS can serve as a model of accelerated VSMC aging and aid in further advances in treating CVD.
Conflict of interestsNo conflicts of interest.
We acknowledge Víctor Quesada for helpful comments. RMN is supported by the Ministerio de Educación, Cultura y Deporte (pre-doctoral contract FPU16/05027). MRH is supported by the Ministerio de Ciencia e Innovación (Juan de la Cierva-Incorporación post-doctoral contract IJC2019-040798-I).