




The establishment of the intestinal microbiota in newborns is a critical period with possible long-term consequences for human health. In this research, the development of the fecal microbiota of a group of exclusively breastfed neonates living in low socio-economic conditions in the city of São Paulo, Brazil, during the first month of life, was studied.
METHODS:Fecal samples were collected from ten neonates on the second, seventh, and 30th days after birth. One of the neonates underwent antibiotic therapy. Molecular techniques were used for analysis; DNA was extracted from the samples, and 16S rRNA libraries were sequenced and phylogenetically analyzed after construction. A real-time polymerase chain reaction (PCR) was performed on the samples taken from the 30th day to amplify DNA from Bifidobacterium sp.
RESULTS:The primary phylogenetic groups identified in the samples were Escherichia and Clostridium. Staphylococcus was identified at a low rate. Bifidobacterium sp. was detected in all of the samples collected on the 30th day. In the child who received antibiotics, a reduction in anaerobes and Escherichia, which was associated with an overgrowth of Klebsiella, was observed throughout the experimental period.
CONCLUSION:The observed pattern of Escherichia predominance and reduced Staphylococcus colonization is in contrast with the patterns observed in neonates living in developed countries.
Microbial colonization of the gastrointestinal tract begins immediately after birth and is carried out by microbes that are derived from the mother and the environment. The gut, during colonization, has classically been described as being initially dominated by a range of facultative bacteria, such as Enterobacteriaceae, Streptococcus, and Staphylococcus. As the available oxygen is consumed, strict anaerobes, including Bifidobacterium, Bacteroides, and Clostridium (1), proliferate.
The course of microbial colonization of the newborn gut is determined by a complex interaction of factors, including the mode of delivery, feeding practice, and the bacterial load in the environment (1). In developed countries decades ago, the first facultative anaerobic colonizers were from the family Enterobacteriaceae, derived from the mother and the hospital environment (2,3). Recently, a shift from this pattern was observed and described as a diminished initial colonization by enterobacteria and high rates of colonization by Staphylococcus (4–6), which was probably derived from the skin of the parents (6). It has been suggested that this change correlates with the stricter hygiene measures applied during and after labor in developed countries (4,5,7-9). In developing countries, however, ideal hygiene control has not yet been achieved, and repercussions in early gut colonization are expected. Initial studies conducted in developing countries observed that neonates were soon colonized by Escherichia coli and other Enterobacteriaceae. Researchers observed that babies harbored a large number of bacterial species, and they had a rapid turnover of bacterial strains in their microbiota resulting from high bacterial exposure in the environment (7,8). A clear understanding of the colonization process of neonates guts in developing countries at the present time is somewhat difficult, as the few available studies were mostly performed more than a decade ago using culture techniques (7,8).
Studies on the microbial colonization of the gut performed using culture techniques may be limited because some bacteria are believed to be unculturable (10). Molecular techniques are able to overcome this limitation because they permit microbes to be identified from the genetic material present in the sample, with no dependence on cultivation. The techniques are mostly based on the study of 16S rRNA, the bacterial small ribosomal subunit RNA. The 16S rRNA contains regions of nucleotide sequences that are highly conserved across the bacterial world, interspersed by variable and hypervariable regions that allow for phylogenetic analyses of the microorganisms (10).
Colonization studies performed in developed countries using molecular methods have permitted more detailed and dynamic analyses of the newborn gut colonization process. Some molecular studies showed colonization patterns similar to those that have been classically described, with the earliest colonizers identified as aerobes, including Staphylococcus, Streptococcus, and Enterobacteriaceae, and later colonizers tending to be strict anaerobes, such as Eubacterium and Clostridia (11). Others observed divergent initial bacterial colonization events, such as the predominance of the anaerobic species Clostridium in the first days of life (12) and the important participation of previously unidentified bacterial groups, such as the anaerobe Ruminococcus (12,13).
Different molecular techniques can be applied to the study of intestinal microbiota, depending on the question being addressed. When the objective is to explore the biodiversity of a sample, construction of a 16S rRNA library can be used (10). Unfortunately, this process may suffer limitations, especially related to the PCR stage (14). The valuable information obtained by the construction of a 16S rRNA library fully justifies its potential pitfalls, but it is important to analyze the results in a judicious manner (15).
This study is the first to use a molecular approach to analyze the gut colonization process of a group of exclusively breastfed neonates born in São Paulo, Brazil, and living in a low socio-economic environment. In one child, the microbial events associated with the use of an antibiotic were also assessed.
MATERIALS AND METHODSStudy group and sample collectionThe study was carried out from June through November of 2007, in the city of São Paulo, Brazil. The study group consisted of ten newborns. All of the mothers underwent prenatal care at a health center at the University of São Paulo. They were well nourished and healthy, and they had not taken any antibiotics during the last month of gestation.
The babies were vaginally delivered at the University Hospital of the University of São Paulo (HU-USP). The newborns had uneventful births and had no associated morbidities; they were full-term and had adequate birth weights. After delivery, they remained in the hospital for 48 hours without incident. During the hospital stay, they did not receive any medication or supplemental fluids. Exclusive breastfeeding was established from birth and continued throughout the research period.
The families belonged to low socio-economic communities. In general, the parents had little schooling and low incomes; families lived in multi-habitation homes, and some of them lived in slums. These communities had electricity and running water, although the access to water in each home varied. The sewer system was inadequate. Only families who lived in communities located in the neighborhoods near the HU-USP and who were referred to this center for medical follow-up were enrolled in the study.
Three fecal samples were collected from each newborn. The first was collected on the 2nd day of life, while the newborn was still in the hospital. The second and the third samples were collected at home (by the mother) on the 7th and 30th days after birth, respectively. A medical appointment was scheduled for these time periods (the 7th and 30th days). The mothers were instructed to collect the fecal sample immediately after elimination with a standardized spoon, to place it in a sterile plastic container, and to keep it in a freezer (-4oC) until the appointment, which occurred later on the same day. Samples were transported inside a polystyrene container filled with ice. Despite their low socio-economic status, all of the families possessed a freezer coupled to a refrigerator and were capable of producing ice for this transport. Because of their geographic proximity, they took less than one hour to deliver the samples to the hospital.
One of the ten children who participated in the study required the use of a 10-day oral cephalexin treatment because of a skin infection that began on the 7th day of life. No 7th-day fecal sample was collected from this child. The 2nd- and 30th-day samples were collected, and the microbial profile of this child is described separately from those of the other children in the group.
DNA extraction and 16S rRNA amplificationDNA was extracted from stools using the QIAamp DNA Stool Mini-Kit (Qiagen, Canada) according to the manufacturer's instructions. Two bacteria-specific primers, 27F (5′AGA GTT TGA TCC TGG CTC AG 3′) and 1492R (5′ GGT TAC CTT GTT ACG ACT T 3′), were used to amplify the 16S rRNA gene coding region (16). Amplification reactions were performed in a total volume of 100 µL containing 180 ng of DNA that was extracted from each fecal sample. The PCR amplifications were performed using the following procedure: 94°C for 5 min, followed by 35 cycles of 94°C for 45 sec, 62°C for 1 min, 68°C for 2 min, and a final extension period of 68°C for 10 min. The amplified 16S rRNA was purified using the UltraClean PCR kit (Invitrogen, USA).
Cloning of the 16S rRNA libraryThe purified PCR products were ligated into a pCR®2.1 TOPO plasmid vector and then transformed into competent E. coli cells using an Original TA Cloning Kit (Invitrogen, USA). 16S rRNA libraries were constructed for each time point (2nd, 7th, and 30th days). After the bacterial transformation, approximately 90 clones from each child at each time point were selected, and their lysates were used as the template in an amplification reaction using the M13 primers supplied by the cloning kit.
DNA sequencing and phylogenetic analysisThe amplicons obtained as described above were purified using the UltraClean PCR kit. The products were analyzed using an automated MegaBACE 1000 DNA Analysis System (GE Healthcare, USA) and Cimarron 3.12 Base Caller (Cimarron Software Inc., USA), and the sequencing reactions were performed using the T7 primer supplied by the cloning kit. Sequencing quality was analyzed using the PRED17 online software, and nucleotides with a Phred score of less than 10 were discarded. Qualified sequences were manually edited using the BioEdit software, v7.0.5.3 (18) to remove the vector and primer sequences. Chimeric artifacts were checked by the Chimera Slayer algorithm in the Mothur program, v1.13.0 (19) using a Silva aligned database as a reference. The phylogenetic affiliations of the resulting sequences (approximately 450 bp and larger) were determined using the Classifier program of the Ribosomal Database Project (RDP) (20) and were confirmed by BLASTn comparison using the NCBI GenBank database.
The Mothur program was used to perform the rarefaction analysis and estimation of the microbial diversity using operational taxonomic unit (OTU)-based approaches (ACE and Chao1 richness estimators, Shannon and Simpson diversity indices) and hypothesis-based approaches (Libshuff statistics and UniFrac distance comparisons) (19). The sequences were aligned using the Silva 102 SSU reference database as a template (21), and a Phylip distance matrix file was generated to cluster the sequences into OTUs. The diversity analyses at the family and genera levels were based on definitions of OTUs with at least 90 and 95% sequence similarity, respectively. A Venn diagram was also drawn to show the number of shared and unique OTUs among each sample. Phylogenetic trees were constructed using the ARB software (22) with the neighbor-joining method and Felsenstein correction. A phylogenetic tree with only the non-chimeric clone sequences was subjected to the weighted and unweighted UniFrac tests, which tested the hypothesis that a group of sequences (that is, branches of a phylogenetic tree) may be unique to any community.
Data presentationThe data from nine of the ten neonates in the study, who progressed without incidents throughout the research period, were analyzed as a group (Group data). The data from the other neonate, who had received an antibiotic, were analyzed separately (Antibiotic exposure data).
The relative abundance of bacterial genus encountered in the fecal molecular analysis was calculated as the percentage of sequences obtained for a specific bacterial genus in relation to the total number of sequences recovered at each time point. The distribution of the abundances of each genus was graphically presented. The prevalence was calculated as the percentage of neonates in the group that harbored a specific bacterial genus.
Real-time PCR for BifidobacteriaBifidobacterium was not detected in the fecal samples using the 16S rRNA library technique. Therefore, a real-time PCR reaction for Bifidobacterium spp. was conducted on the 30th-day fecal samples for further investigation. The primers and probe were the same as those validated by Penders et al. (23). The amplification reaction was conducted in a total volume of 50 µL, containing 1x TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA), 300 nmol/L of both primers, 150 nmol/L TaqMan probe, and 0.2 ng of the purified target DNA. The amplification reactions were performed using the following procedure: 50°C for 2 min, 95°C for 10 min, and 42 cycles each of 95°C for 15 seconds and 60°C for 1 min. PCR products were detected with an Applied Biosystems Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA).
To determine the rrn copy number in the fecal samples, standard curves were calculated by correlating the rrn gene copy number and the threshold cycle (Ct) values in a ten-fold serial dilution of Bifidobacterium animalis subsp. lactis HN019. The data quantification was expressed as the range of the values encountered in the group and the mean.
Nucleotide sequence accession numberThe nucleotide sequences reported in this paper will appear in the DDBJ, EMBL, and GenBank nucleotide sequence databases with the following accession numbers: 2nd day - GU145617 to GU146046, 7th day - GU131996 to GU132306, and 30th day - GU229292 to GU229644.
Ethical considerationThis study was approved by the Ethics Committee of the HU-USP (under registration number 574/05). The mothers of all of the infants enrolled in the research signed an informed consent form.
RESULTSData related to the individualsThe studied neonates had a mean gestational age of 39.6±1.6 weeks (range 37.3 to 41.6 weeks) and a mean birth weight of 3,328±388 g (range 2,725 to 3,560 g). Table 1 displays the characteristics of the neonates and their antibiotic use.
Data showing the general characteristics and antibiotic use of the neonates.
| Gestational age(weeks) | Birth weight | Gender | Antibiotic use | |
|---|---|---|---|---|
| 1 | 40.5 | 3.560 g | F | No |
| 2 | 40.1 | 2.870 g | F | No |
| 3 | 41.1 | 2.765 g | F | Yes* |
| 4 | 41.6 | 3.725 g | M | No |
| 5 | 37.6 | 3.030 g | M | No |
| 6 | 39.3 | 3.300 g | F | No |
| 7 | 37.3 | 3.210 g | M | No |
| 8 | 40 | 3.290 g | M | No |
| 9 | 40.6 | 3.520 g | M | No |
| 10 | 38.1 | 4.015 g | M | No |
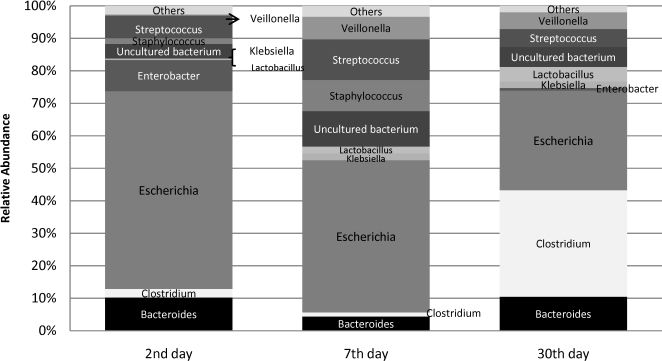
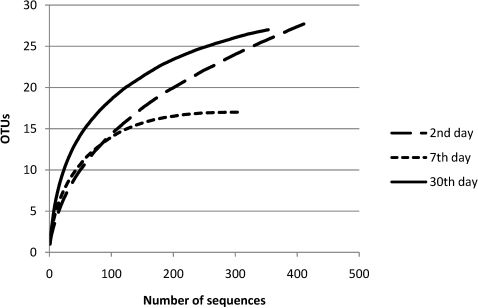
Approximately 2,610 clones were obtained to construct the 16S rRNA gene library. A total of 2,419 clones were sequenced. After excluding the low-quality or chimeric sequences, 1,765 sequences, each encompassing approximately 450 bp from the 5′ end of the 16S rRNA gene (Table 2), were gathered and used to analyze the prevalence and relative abundance of microbes at each time point (Table 3, Figures 1 and 2). Rarefaction curves and the diversity estimation were constructed using only the forward-aligned sequences (1,077 sequences, Table 2). The rarefaction curves (Figure 3) illustrate the number of sequences that were acquired and the number of OTUs that these sequences represent at a species-level cutoff (≥97% similarity). Although the clone library coverage was high for all of the samples (>96%, Table 2), which indicates that the sampling effort was sufficient to cover the community diversity, only the 7th-day sample rarefaction curve showed a slight plateau (Figure 3). Nevertheless, the slope of the 7th-day curve was shallower than the slope of the curves obtained for the 2nd and 30th days, which suggests that greater numbers of OTUs are expected in these two libraries.
Richness estimators, diversity indices, and coverage of bacterial 16S rRNA gene sequences from 2-, 7-, and 30-day-old neonates (the OTU was defined as a ≥95% similarity cutoff).
| Sample | No. of sequencesa | OTUs | Richness estimators | Diversity indices | Coverage (%) | ||
|---|---|---|---|---|---|---|---|
| ACE | Chao1 | Simpson | Shannon | ||||
| 2-day-old | 421 | 28 | 67 | 48 | 0.440 | 1.516 | 96.9 |
| 7-day-old | 304 | 17 | 18 | 17 | 0.246 | 1.891 | 99.9 |
| 30-day-old | 352 | 27 | 31 | 29 | 0.132 | 2.437 | 98.5 |
a – the number of forward-oriented clone inserts (5′-3′) after excluding those of weak quality and suspected chimeras.
Relative abundances and prevalences of bacterial genera identified in fecal samples of nine neonates throughout the first month of life.
| Bacterial genus | 2nd day (n = 9) | 7th day (n = 9) | 30th day (n = 9) | |||
|---|---|---|---|---|---|---|
| P* | RA** | P* | RA** | P* | RA** | |
| % | % | % | % | % | % | |
| Bacteroides | 33 | 10 | 22 | 4 | 33 | 10 |
| Clostridium | 44 | 3 | 22 | 1 | 56 | 33 |
| Escherichia | 100 | 61 | 78 | 47 | 67 | 31 |
| Enterobacter | 44 | 9 | 0 | 0 | 33 | 1 |
| Klebsiella | 22 | 1 | 22 | 2 | 44 | 2 |
| Lactobacillus | 11 | 1 | 22 | 3 | 33 | 5 |
| Staphylococcus | 22 | 2 | 33 | 10 | 0 | 0 |
| Streptococcus | 56 | 7 | 44 | 13 | 44 | 6 |
| Uncultured Bacteria | 60 | 3 | 78 | 10 | 80 | 5 |
| Veillonella | 22 | 1 | 44 | 7 | 44 | 5 |
| Others | 25 | 2 | 44 | 3 | 44 | 2 |



Richness estimates obtained by both ACE and Chao1 were in agreement, demonstrating that the microbiota of the 2nd-day group had a greater richness than those of the other time points (Table 2). Interestingly, both the Simpson and Shannon indices showed the lowest diversity in the 2nd-day group and the highest diversity in the 30th-day group (Table 2). In fact, the large number of clones similar to Escherichia found in the 2nd-day group of fecal material accounted for the dominance measured by the diversity indices, as shown in the compositional analysis. On the other hand, the microbiota of the 30-day-old neonates had a different community structure, composed of a smaller number of bacteria distributed more equally among communities; that is, these communities appeared less dominant. A detailed analysis of the OTUs found in each sample group (Table 2) is shown in a Venn diagram (Figure 4). The 2nd-day sample group had the greatest number of unique sequences (14 OTUs), followed by the 30th-day (10 OTUs), and 7th-day (4 OTUs) sample groups. Nevertheless, approximately 12.7% of the gut species (6 OTUs) were shared among all time points, which suggests that these genera were present throughout the first month of life.
.")
Analysis by hypothesis-based approaches, which tests if the microbial communities have the same structure by chance, revealed significant differences among each group. The Libshuff test resulted in significance values (<0.0001 for each sample comparison) below the critical threshold (i.e., 0.05/6 = 0.00833), which indicates that each neonate microbial community had a different structure. In agreement with these results, both the unweighted and weighted UniFrac tests indicated that each sample had a different community structure (significance values <0.001 for each sample and a critical threshold of 0.05/3 = 0.01666 for each test).
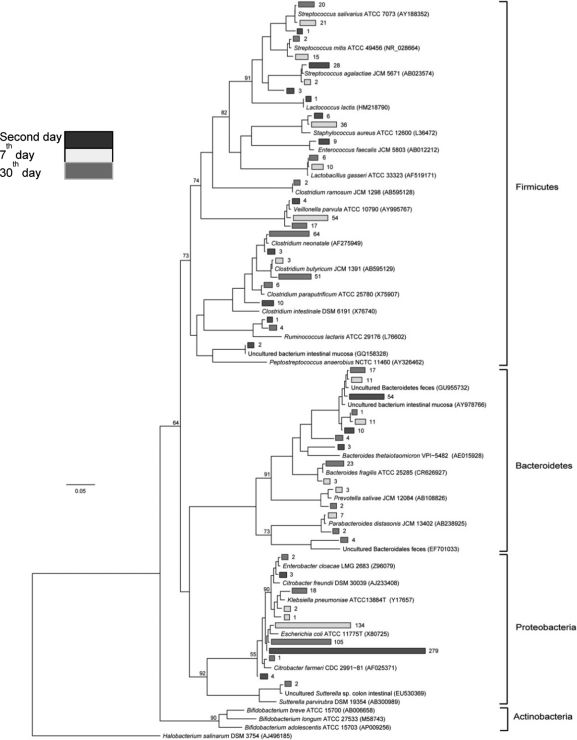
Community composition analysisA detailed, genus-level analysis of the OTU affiliations, as determined by the RDP Classifier and BLASTn matches, is given in Figure 1. All groups of OTUs revealed members of the phyla Firmicutes, Bacteroidetes, and Proteobacteria (Figure 5).

Phylogenetic relationships among the OTUs detected in newborn fecal samples. Black branches represent the 2-day-old samples, white branches represent 7-day-old samples and gray branches represent 30-day-old samples. Reference sequences are indicated by their taxonomic names, and uncultured clones are followed by their GenBank accession numbers.
2nd-day analysis. A total of 606 sequences were obtained from nine neonate fecal samples (Tables 2 and 3). In the initial 16S rRNA library profile, Escherichia predominated (Figure 1), with a relative abundance of 61% and a prevalence of 100% (Table 3). Other facultative anaerobes were observed, including gram-negative enterobacteria, namely, Enterobacter (44% prevalence, 9% relative abundance) and Klebsiella (22% prevalence, 1% relative abundance), and gram-positive cocci, namely, Streptococcus (56% prevalence, 7% relative abundance) and Staphylococcus (22% prevalence, 2% relative abundance). The strict anaerobes, Bacteroides (33% prevalence, 10% relative abundance) and Clostridium (44% prevalence, 3% relative abundance), were already present in these early days of life.
7th-day analysis. A total of 478 sequences were obtained from nine neonate fecal samples (Tables 2 and 3). By this time point, Escherichia had become less predominant, but it still accounted for the highest number of sequences as it was responsible for 47% of the sequences obtained and occurred in 78% of the neonates. Other enterobacteria (Klebsiella and Enterobacter) were observed in individual neonates at a low abundance. By the 7th day, the facultative anaerobe Streptococcus was still identified in a larger proportion of neonates than Staphylococcus (prevalence of 44% versus 33%), but the two species were similarly abundant (13% versus 10%). The anaerobic components, Bacteroides and Clostridium, were infrequently detected; both anaerobes were identified in 22% of the neonates, with relative abundances of 4% and 1%, respectively. Veillonella was more frequently identified in this analysis (prevalence of 44% and relative abundance of 7%). The anaerobic lactic acid bacterium Lactobacillus was detected in 22% of the samples at a low relative abundance (3%). Other microorganisms that were less frequently identified included Acinetobacter and Parabacteroides, which were detected in different neonates. At this time point, a number of OTUs with genetic similarity to the clones designated “uncultured bacterium” in the data bank were detected in 78% of the samples, and this prevalence was higher than those at other time points.
30th-day analysis. A total of 506 sequences were obtained from nine neonate fecal samples (Tables 2 and 3). Overall, a more uniform distribution of abundances was observed (Figure 1). Clostridium joined Escherichia as the main genera encountered at this time point. Both were of similar relative abundance (Clostridium 33% and Escherichia 31%), and both were detected in a significant number of neonates (the prevalence of Clostridium was 56% and that of Escherichia was 67%). Other enterobacteria were found at a low abundance: Klebsiella was identified in 44% of the neonates with a relative abundance of 2%, and Enterobacter was detected in 33% of the neonates with a relative abundance of 1%. Streptococcus could be identified in 44% of the neonates with a relative abundance of 6%, but sequences related to Staphylococcus were not identified at this time point. The anaerobic components dominated this library for this time point (in total, they were responsible for 58% of the sequences). Apart from Clostridium, Bacteroides could be detected in 33% of the neonates with a relative abundance of 10%; Veillonella was more predominant, being detected in 44% of the neonates with a relative abundance of 5%; and Lactobacillus was moderately better represented in this 30th-day library than in the libraries from earlier time points, being observed in 33% of the neonates with a relative abundance of 5%. Other microorganisms that could be identified were Acinetobacter baumanii, Citrobacter, Enterococcus, and Prevotella.
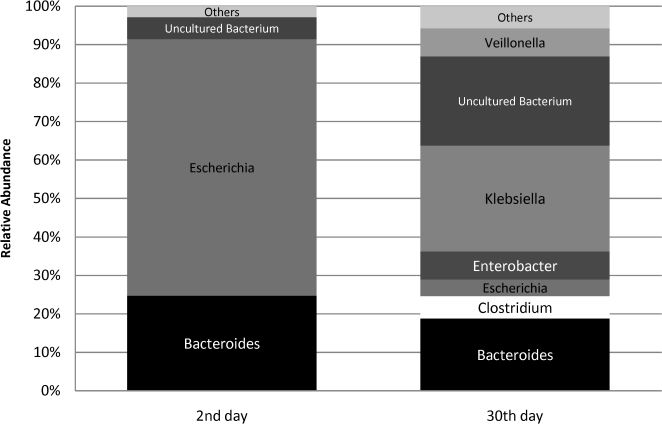
Antibiotic exposure data2nd-day analysis. A total of 106 sequences were obtained from the second-day fecal sample of the child who later underwent antibiotic treatment. The bacterial profile of the 16S rRNA library at this initial time point was similar to those observed for the remainder of the group. Escherichia was the main phylogenetic group identified (Figure 2), responsible for 67% of the sequences (versus 64% encountered in the non-antibiotic group). As for the anaerobic components, Bacteroides was also the major anaerobe identified, but Clostridium was not detected (Figure 2). A small percentage of sequences (6%) were attributed to uncultured bacteria.
7th-day analysis. The 7th-day fecal sample was not collected as the child could not attend the appointment on this day.
30th-day analysis. Sixty-nine sequences were obtained from the 30th-day fecal sample. The microbial profile after antibiotic use showed a significant decline in the relative abundance of Escherichia, which accounted for only 4% of the sequences. Instead, Klebsiella was responsible for a greater proportion of sequences obtained (28%). Anaerobic components were responsible for 35% of the clones, but they did not represent the major phylogenetic group of the library, as was observed in the remainder of the group at this time point. Bacteroides was the main anaerobe identified, and Clostridium was detected at low rates. The proportion of uncultured microorganisms increased; they were detected at a relative abundance of 23%.
Real-time PCR of BifidobacteriaReal-time PCR of Bifidobacterium detected this microorganism in all neonate stools from the 30th day, even though it was absent from the 16S rRNA library. Quantification revealed counts that ranged from 5.61×1010 to 1.46×1012 copies per µg of DNA (median value of 7.20x1011 copies per µg of DNA) in the group of children that did not receive antibiotics and 2.95×1011 copies per µg of DNA in the feces of the child who underwent antibiotic treatment (Table 4).
Bifidobacterium counts obtained through real-time PCR in the 30-day-old samples.
| Infant# | rrn copy number (±SD) |
|---|---|
| 1 | 7.38×1011 (1.01) |
| 2 | 3.37×1011 (4.98) |
| 3 | 2.95×1011 (7.51) |
| 4 | 8.44×1011 (4.56) |
| 5 | 1.46×1012 (9.67) |
| 6 | 1.02×1012 (3.37) |
| 7 | 8.21×1011 (1.95) |
| 8 | 6.73×1011 (8,55) |
| 9 | 9.49×1011 (1.45) |
| 10 | 5.61×1010 (2.68) |
SD: standard deviation of the means of experimental results.
The study population represented a healthy group of neonates who were not submitted to any medical intervention and who were exposed to the average environmental conditions present in poor communities. These characteristics must be emphasized because some published data are from heterogeneous groups of neonates, which complicates interpretation of the observed outcomes. Data from the one baby who received an antibiotic were analyzed separately because this intervention had the potential to affect normal microbial establishment. All babies were vaginally delivered and exclusively breastfed, two factors that strongly influence the colonization process.
Taking into account this scenario and the need to fully describe the microbial characteristics of this group of Brazilian neonates, the 16S rRNA clone library methodology was applied, rather than high-throughput sequencing methods (24,25). This method allowed the characterization of the most abundant organisms, despite the fact that this approach may result in a lower assessment of the bacterial diversity.
Richness estimates obtained by both the ACE and Chao1 indices and diversity estimates obtained by both the Simpson and Shannon indices showed the lowest diversity in the second-day group and the highest diversity in the 30th-day group. These results demonstrate that the neonates' microbiota shifted from a simple to a more complex bacterial profile. It has previously been demonstrated that the microbiota of infants grow in complexity of microbial communities until a stable, adult-type microbiota is achieved (24). Each neonate microbial community had a different structure and was composed of members of the phyla Firmicutes, Bacteroidetes, and Proteobacteria. This characteristic of an individual microbiota pattern, or inter-individual variability, has been described for the first days of life (24,25). This variability must reflect the consequences of the selective forces that act inside each individual (e.g. number of bacterial receptors in the gut and each ones particular gastrointestinal physiology, gastric acid, peristalsis, bile flow), the impact of the environment and the inter-bacterial arrangements (1). The importance of each individual microbe to the host must be better understood. Although primordial microbial communities are vulnerable to the influences of external environment, ecological stability is achieved by approximately two years of age. Furthermore, although individuals are still exposed to environmental microbes after this age, further attempts to interfere with the composition of the microbial community will produce only transient effects (26).
Although individual characteristics were noted, a tendency toward a pattern of microbial colonization existed, and the findings were further considered in a group manner. When the bacterial profiles of the neonates' fecal microbiota were analyzed as a group, Escherichia stood out as a major bacterial group at all time points. In the second-day samples, clones derived from Escherichia could be identified in all of the neonates; this microorganism accounted for 61% of the clones obtained. In subsequent samples, its relative abundance declined somewhat. However, even though other bacterial groups had become more abundant in the 30th-day samples, Escherichia was one of the major phylogenetic groups in the library. This high degree of enterobacteria colonization observed throughout the first month of life in these Brazilian neonates was observed in other studies conducted in newborns living in developing countries (7,9,27) and is possibly related to a high degree of environmental exposure and contamination. In a study that analyzed the colonization pattern of Pakistani babies, a massive colonization by enterobacteria was found to have already occurred in the first days of life, and this colonization persisted throughout the first few months (7). The environment was identified as the main source of contamination because even though all of the infants had been vaginally delivered at home, fewer than 50% of the Escherichia strains matched maternal strains, and most of the strains could be found in the immediate home environment (7).
This high rate of enterobacteria colonization, particularly in the first days of life, was also described in initial studies that analyzed the microbiota colonization pattern of neonates living in developed countries (2,28). However, in later years, some authors have asserted that this initial enterobacteria predominance would have greatly declined in neonates living in developed countries (4,5,29,30). Nowrouzian et al. (30) observed that in a group of Swedish neonates, fewer than 50% were colonized by Escherichia by one week of age. This change in colonization may have occurred as a result of development and improved hygiene practices that alter the diversity and abundance of the microbes to which the neonates are exposed.
Moreover, in association with this phenomenon, an increased colonization rate by Staphylococcus was also detected (4–6). The source of the Staphylococcus found among infants' intestinal microbiota was predominantly the parents' skin and not the environment (6). It has been suggested that in the absence of enterobacterial competition, Staphylococcus proliferation would be favored (4). Interestingly, in the present study, clones derived from Staphylococcus were identified in few individuals and at low abundance. It is possible that the opposite to the Staphylococcus favoring phenomena described before has occurred in the microbiota of these Brazilian neonates, whereby the maintenance of a large enterobacterial population inhibited Staphylococcus proliferation. The high prevalence and relative abundance of clones attributed to Clostridium in the 30th-day analysis were also of interest. Clostridium clones could be identified in 60% of the neonates. This finding is not usually reported in exclusively breastfed neonates, in whom low rates of colonization have been described (23,31).
In 1,032 fecal samples from one-month-old Dutch infants, Clostridium was detected in only 21% of those who were breastfed and only at low levels (4.5 log10 CFU) (23). Newborn colonization by Clostridium is attributed to a lesser extent to mother-to-child transmission and is mainly caused by environmental contamination and transfer via contaminated hands (32). The Clostridium colonization frequency has been correlated with the degree of contamination by this microorganism in the environment to which the child is exposed (33). It is possible that the neonates enrolled in this investigation were exposed to a high load of Clostridium in their domestic surroundings and that poor hygiene conditions favored contamination. Considering the fact that these neonates were exclusively breastfed, and knowing that being fed by human milk creates an intestinal milieu that is not favorable for Clostridium proliferation (34), higher colonization would be expected if they had not been breastfed.
This microbial pattern of persistently high levels of Escherichia without over-proliferation of Staphylococcus and relatively high rates of Clostridium colonization, unlike those reported in developed countries (4), might reflect the impact of environmental contamination on intestinal colonization. The effect of the environmental microbial load to which an individual is exposed, and the microbial composition of this load, on the development of the immune system is debated in the “hygiene hypothesis” (35). According to the hygiene hypothesis, the stricter hygiene practices adopted in developed countries may modify the initial microbial exposure, with a negative impact upon immune regulation, possibly leading to the greater incidences of allergic and autoimmune diseases observed in these countries (35).
Studies have provided evidence for the role of the initial intestinal colonizers in the development of the naïve immune system and shown that the sequence of bacterial events that occurs during colonization of the gastrointestinal tract may affect the host's future health (36). The challenge is to fully understand which microbial events (their timing and composition) are able to modify the host's future health and whether interventions in events that occur in the beginnings of life, could favor the establishment of a more beneficial microbiota.
An intriguing feature of the constructed libraries was that no Bifidobacterium could be identified at any of the analyzed time points. Although some studies (37) have reported low rates of Bifidobacterium even in breastfed neonates, high Bifidobacterium levels are expected in exclusively breastfed infants owing to particular features (acid profile, iron content, and bifidogenic factors) of human milk that favor Bifidobacterium growth to the detriment of other microorganisms (34), and many studies have provided evidence for the differential high levels of Bifidobacterium colonization in breastfed neonates (23,25,29). Other studies have concluded that the finding of appropriate levels of Bifidobacterium might be related to methodological aspects of each particular study and that the particular approach used can alter whether high or low levels of Bifidobacterium are detected (25).
In view of this unexpected result, another molecular approach was used to investigate, in a more sensitive and specific manner, the occurrence of Bifidobacterium in the 30th-day samples. A specific probe and primers for Bifidobacterium sp. were used in a real-time PCR reaction, which allowed the detection and quantification of such bacteria.
This technique allowed detection of Bifidobacterium in the samples from all of the children at one month of age. Quantification of the samples revealed average counts of 7.20x1011 copies per µg of DNA. Thus, frequent colonization and high population levels of Bifidobacterium were in fact detected in these breastfed neonates, in accordance with other studies (23,29).
In a study that used culture techniques to analyze the difference in Bifidobacterium and Lactobacilli populations among Brazilian students from two different socioeconomic groups, de Mello et al. (38 )observed lower numbers of Bifidobacterium and Lactobacilli in the children living in slums compared with those who attended private schools. The authors suggested that these microbial populations might have been affected by the unfavorable slum environment. A possible explanation for the divergent finding of low Bifidobacterium levels in children exposed to poor environment in Brazil observed by de Mello GQ et al. (38) in Brazilian students living in slums could be the fact that those children were older and had thus been subjected to environmental aggressions over a longer period of time, in the absence of the protective and bifidogenic effect of breastfeeding.
The inability of our molecular approach to amplify Bifidobacterium DNA from the samples can be explained in view of the inevitable bias involved in PCR-dependent methodologies (14). Bifidobacterium was also not identified in some other studies that used the 16S rRNA library methodology to analyze intestinal microbiota (16,24); therefore, the ability of this molecular technique to adequately detect the Bifidobacterium genus must be questioned.
The ecological structure that forms inside the gut can be disrupted by the use of antibiotics, which can suppress susceptible bacterial groups and favor the overgrowth of others (8). The future impact of this initial microbial imbalance is not clear, but it is possible that the use of antibiotics early in life may affect the immune regulation process (39). The impact of antibiotic use was investigated through the follow-up of one child who required the use of cephalosporin during the research period. It is known that different antibiotics affect microbiota components in different ways. Approximately ten days after antibiotic use, the microbiota profile of the antibiotic-treated child revealed changes in the bacterial communities compared with the remainder of the group. These differences could represent an effect of the antibiotic use. A significant reduction in the relative abundance of Escherichia was observed. This microorganism was found at levels 7.5 times lower than those encountered in the remaining children. In turn, Klebsiella was found at 14-fold higher levels. This change in the enterobacterial profile, with a replacement of Escherichia by an overgrowth of Klebsiella, has been highlighted by other researchers who analyzed the impact of similar antibiotic groups on the composition of children's microbiota (8). The prevalence of anaerobic microorganisms may also have been reduced because of the antibiotic exposure. In contrast with the findings for the non-antibiotic user group, anaerobic bacteria were not detected as a predominant group over the aerobic components. This effect of antibiotics on the anaerobic bacteria components was also described by previous researchers, who reported a significant impact of antibiotics in reducing Bifidobacterium levels (8). The effects of alterations in Bifidobacterium levels could not be clearly established, as Bifidobacterium were encountered in the 30th-day samples at a level somewhat lower than that of the group average, although still within the group range, possibly indicating a quick reestablishment favored by breast milk.
Although described in only one child, a disruption of the initial microbial colonization process by the use of an antibiotic is suggested. The long-term impact of this intervention should be taken into account when analyzing the indication for antibiotics early in life because the microbiota composition may change definitively after antibiotic use. Some studies have shown a tendency for microbiota composition to be restored after antibiotic use, but with some permanent changes (40). The microbiota profile of the antibiotic-treated child revealed changes in the bacterial communities during the first month of life. In the following months, this profile showed a turnover of Escherichia (resulting in 30% of the microbial population) but also became more diverse than the profiles of the children who were not given antibiotics (data not shown), suggesting that the microbial profile was not completely restored.
This molecular approach permitted the analysis of fecal microbiota establishment in a group of neonates from São Paulo, Brazil. The observed bacterial predominance of Escherichia and Clostridium may be attributable to a highly contaminated environment, and neonate contamination may have been favored by hygiene habits. The observed pattern of predominance by Escherichia in the first days of life and the low rates of colonization by Staphylococcus differ from the colonization patterns described in developed countries. It is possible that a greater knowledge of these differences might help us to understand the ways in which intestinal microbes can favor adequate regulation of the immune system.
AUTHOR CONTRIBUTIONSBrandt K conceived the study, performed children selection, sample collection and prepared the manuscript. Taddei CR designed the study, performed the experiments and data analysis and prepared the manuscript. Takagi EH contributed to the study design, execution of experiments, data analysis and bioinformatics results. Oliveira FF contributed to the execution of experiments and data analysis. Irino I contributed to the execution of experiments. Duarte RTD contributed to the data analysis and bioinformatics results. Martinez MB designed the study, performed the data analysis and prepared the manuscript. Carneiro-Sampaio M conceived the original study, contributed to the data analysis and supervised the preparation of the manuscript.
We thank Professor Luiz R. Trabulsi (in memorian) for having idealized the original study with Carneiro-Sampaio M.
No potential conflict of interest was reported.