







Traumatic brain injury (TBI) is associated with hypopituitarism with a variable incidence, depending on the time and methods used to diagnosis, and on factors related to the trauma, such as its severity, its anatomical location and the drugs used in the acute phase. The pituitary gland can be damaged directly by the impact or secondary to factors such as ischemia, inflammation, excitotoxicity or immunity. In acute phases ACTH deficiency is the most relevant, since failure to detect and treat it can compromise the patient's life. Clinical manifestations are typical of each hormone deficient axes, although the combination hypopituitarism-trauma has been associated with cognitive deterioration, worse metabolic profile and greater impairment of quality of life. One of the clinical challenges is to determine which patients benefit from a systematic hormonal evaluation, and therefore from hormone replacement, and what is the appropriate time to do so and the most suitable diagnostic methods.
El traumatismo craneoencefálico (TCE) se asocia con hipopituitarismo con una incidencia variable, dependiendo del momento y los métodos diagnósticos utilizados y de factores relacionados con el traumatismo, como la gravedad, localización anatómica y los fármacos utilizados la fase aguda tras el mismo. La hipófisis puede dañarse de forma directa por el impacto o de forma secundaria a factores como la isquemia, la inflamación, la excitotoxicidad o inmunidad. En fases agudas detectar el déficit de ACTH es vital. Además de las manifestaciones clínicas típicas del hipopituitarismo, se ha asociado con mayor deterioro cognitivo, peor perfil metabólico y mayor afectación de la calidad de vida. Uno de los retos clínicos es determinar qué pacientes se benefician de una evaluación hormonal sistemática tras sufrir un traumatismo, y por tanto de sustitución hormonal y cuál es el momento y metodo diagnostico apropiado para hacerlo.
Traumatic brain injury (TBI) affects 69 million people yearly, with an incidence in industrialised countries of 200–235 cases/100,000 inhabitants per year. It is the leading cause of death in young people, and in non-fatal cases it is associated with cognitive, behavioural, and social sequelae in the medium and long term, in addition to neuroendocrine dysfunction.1,2
Epidemiology of hypopituitarism after TBIThe first description of hypopituitarism after TBI dates from 1918.3 Many studies have subsequently shown that cranial trauma is associated with pituitary hormone dysfunction. In a series of adult patients with hypopituitarism, TBI was the cause in 1.4% of cases,4 though its current impact is expected to be higher due to greater recognition and research.
The published studies on the prevalence of hypopituitarism after TBI show variable results, probably because of their heterogeneity in terms of trauma severity and diagnostic methodology.5,6 In a meta-analysis of 2007,7 the overall prevalence of hypopituitarism after TBI was 27.5%, and varied by severity of TBI, assessed using the Glasgow Coma Scale. It was 35.3% in severe cases (GCS≤8), 10.9% in moderate cases (GCS 9−12), and 16.8% in mild cases (GCS≥13). Most studies,8,9 but not all10,11 found greater prevalence with greater trauma severity, and most cases considered “mild” required hospitalisation and/or neurosurgical intervention.
The prevalence of hypopituitarism is also determined by the time elapsed after TBI. In a systematic review involving over 5000 patients, the prevalence of hypopituitarism after TBI was 45% (<3 months), 36% (3–12 months), and 32% (>1 year).12 In contrast, prospective studies found a prevalence of up to 50.9% one year after TBI.8,13
GH deficiency, the most common and usually as an isolated deficiency, was detected in 22% of cases, followed by hypogonadism (10.2%), hypocortisolism (10%), and hypothyroidism (6.2%).14 Hormonal changes consistent with GH, gonadotropin, or TSH deficiency represent physiological responses to acute stress that resolve in subsequent assessments. In contrast, ACTH deficiency is clinically significant and may compromise patient life if not treated. In the acute phase of TBI, there are adaptive changes to stress that include an increase in serum cortisol, loss of circadian rhythm, and lack of cortisol suppression after 1mg dexamethasone. Reduced cortisol secretion in acute phases leads to increased morbidity and mortality. ADH deficiency is common in the acute phase and is usually transient.11 Its occurrence is associated with a poor prognosis.15
Pathophysiology of hypopituitarism after TBISeveral pathophysiological mechanisms have been described that are involved in development of hypopituitarism after TBI. Its occurrence is determined by different factors18 (Table 1).
Factors related to occurrence of hypopituitarism after TBI.
| Location of injury | • Pituitary capsule, anterior or posterior pituitary lobe, pituitary stalk, vascular system. |
| • Skull base fractures. | |
| • Visible injury on imaging tests. | |
| Severity of TBI | • Based on GCS score, mild: 13−15 points, moderate: 9−12 points or severe ≤8 points. |
| • Almost 50% have adrenal insufficiency after moderate-to-severe TBI. | |
| Presence of secondary brain damage | • Hypotension, hypoxia, cerebral oedema, haemorrhage, and inflammatory and immune changes (persistent neuroinflammation or autoimmunity). |
| • Diffuse axonal damage. | |
| Drugs in critical illness | • Opioids, phenobarbital, high-dose heparin, or etomidate have been related to adrenal axis changes. |
| Other factors | • Advanced age. |
| • Prolonged ICU stay. |
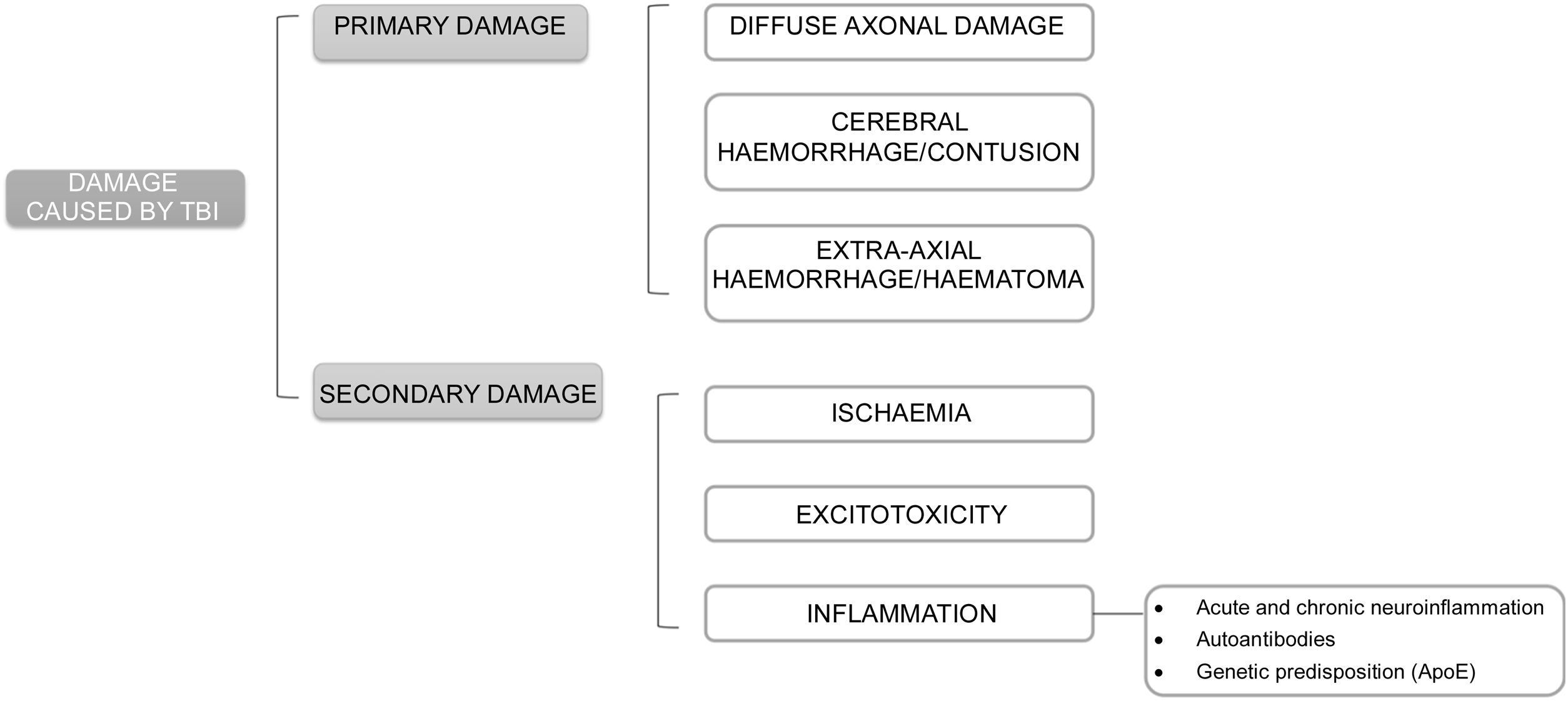
The pathophysiology underlying damage after TBI may be classified as primary and secondary damage.
Primary damage occurs at the time of TBI either by direct effect of external forces transferred to intracranial content or by compression by surrounding structures. It may induce diffuse axonal damage, cerebral bleeding or contusion, and extra-axial bleeding or bruising. Skull base fractures and pituitary haemorrhages are the most likely to induce pituitary damage.
Secondary damage is related to mechanisms triggered after TBI such as ischaemia, excitotoxicity, and inflammation (Fig. 1).
- 1
Ischaemia: it may occur in the setting of vasospasm, vascular damage or microvascular occlusion, and trigger hypoxia, oedema, hypotension or anaemia. The risk of ischaemia is greater in the anterior hypophysis, whose blood supply depends on longer vessels, as compared to the stalk and posterior hypophysis that are nourished by small portal capillaries, less susceptible to damage. This explains the greater frequency of anterior hypopituitarism. Within this, GH and gonadotropin deficiencies are more common after ischaemic damage because these cells are found on the sides of the pituitary gland, nourished by larger vessels that are more susceptible to damage. In contrast, TSH-ACTH deficiencies would be less common because thyrotropic and corticotropic cells are located in the central part of the pituitary gland, vascularised by small, less exposed vessels.
- 2
Excitotoxicity: caused by an abnormal elevation of excitatory neurotransmitters, which are released uncontrollably after TBI. At high concentrations they act on ion channels, altering cell permeability and causing an abnormal flow of electrolytes between the intracellular and extracellular spaces.16
- 3
Inflammation: after a TBI, an acute general inflammatory reaction occurs that can subsequently become chronic. Thus, mild TBIs may cause a transient alteration of the blood-brain barrier allowing a temporary flow of inflammatory molecules and cells through that damaged barrier until it recovers. However, severe or repeated trauma can cause chronic inflammation that can last for months or years.

Another mechanism related to inflammation after TBI is autoimmunity. Following TBI, patients develop an immune response (anti-pituitary or anti-hypothalamic antibodies) after blood-brain barrier disruption and outflow of brain proteins. Such responses have been detected even with chronic mild trauma (boxers) and may persist for up to 5 years after diagnosis. High antibody titres have been related to greater development of hypopituitarism and less recovery and are considered markers and risk factors for pituitary damage. Antihypothalamic antibodies are related to GH and ACTH deficiency, without ADH deficiency, suggesting that they are mainly directed to somatotropic and corticotropic cells and not to the posterior pituitary gland. However, most evidence comes from the same research group, with relatively small samples, non-prospective studies, and no early assessment of pituitary function after trauma. Therefore, more evidence would be needed before incorporating routine antibody measurement.
Genetic predisposition may also be implicated in pituitary dysfunction after TBI. Apolipoprotein E increases after trauma and reduces neuroinflammation and repairs cell membranes. Some ApoE polymorphisms may have prognostic implications. ApoE3 has more marked anti-inflammatory properties and reduces levels of proinflammatory cytokines at the systemic and nervous level, with better results in pituitary damage after TBI, while the ApoE4 polymorphism has been associated with a poorer prognosis.17
Clinical signs and symptomsThe manifestations of hypopituitarism after TBI range from mild and non-specific symptoms (asthenia, anorexia or headache) to severe symptoms that can be life-threatening for the patient and require immediate treatment, such as acute adrenal crisis or severe water-electrolyte disorder. The number of hormone deficiencies, their severity, or the time to treatment will be key to clinical expression of hypopituitarism.
In addition to clinical signs related to hormone deficiencies, this hypopituitarism may be associated with a greater frequency of cognitive, metabolic, and quality of life changes (Table 2) (36).
Clinical signs of hypopituitarism after TBI.
| Classical symptoms of each hormone deficiency | - ACTH: asthenia, anorexia, hypotension, hypoglycaemia, anaemia, hyponatremia. Adrenal crisis. |
| - TSH: asthenia, constipation, weight gain, dry skin, hair loss. | |
| - FSH/LH: decreased libido, weight changes, osteoporosis, feminisation and decreased hair (males), oligoamenorrhea (females). | |
| - GH: fatigue, loss of strength and muscle mass, central fat redistribution, osteoporosis, decreased quality of life, memory impairment. | |
| - ADH: diabetes insipidus. | |
| Neurocognitive changes | - Changes in memory, attention, executive functions, processing speed or even language. |
| Metabolic changes | - Weight and BMI gain, abdominal fat distribution, decreased bone density, blood glucose and lipid profile changes. |
| Poorer quality of life | - Poorer quality of life test score, greater impairment in the physical health, energy, fatigue, emotional well-being or pain domains, as well as general health. |
In the acute phase (first 2 weeks after TBI), there are two key situations due to their life risk and need for immediate treatment: acute adrenal crisis and natremia changes.15
GH and gonadotropin deficiencies are clinically irrelevant in this phase. Central hypothyroidism may contribute to the clinical signs of asthenia, lethargy, and confusion, but it is difficult to discriminate its effect in early stages. Hyperprolactinemia due to stalk compression or stress may cause menstrual changes and sexual dysfunction together with hypogonadism.18
Neurocognitive changesThey are a common sequel after TBI and can affect attention, memory, processing speed, executive functions or more robust functions, such as language and visuospatial construction skills, making it difficult for patients to return to an active work and social life.
Hypopituitarism, trauma-induced damage, or post-traumatic stress syndrome are related to the development of cognitive changes. Although there is clinical overlap between them, it is important to make a correct differential diagnosis, since cognitive symptoms related to hypopituitarism may improve with hormone replacement. Each pituitary hormone deficiency may contribute to the occurrence of neurocognitive clinical signs, but GH has the greatest impact, related to impairment of dendritic, somatic, and neuronal growth, as well as to changes in regulation of brain glucose metabolism in cortical areas related to memory, executive, and intellectual functions. Some studies, but not all, show poorer cognitive results in the presence of GH deficiency.
Hypothyroidism is associated with delayed information processing or short-term memory changes. Hypogonadism in men has been associated with the risk of developing Alzheimer’s disease and treatment with testosterone improves memory. In women, oestrogen replacement has a more conflicting result on cognitive function. Hypocortisolism may lead to mood changes, memory loss, or even overt psychosis.
Metabolic changesThe most common include blood glucose changes, insulin resistance, dyslipidaemia, weight and body mass index (BMI) gain, changes in body composition with abdominal fat distribution, and decreased BMD.19
The factors that promote these metabolic changes after a TBI are summarised in Table 3. Hypothyroidism reduces basal metabolic output, hypogonadism is related to bone and muscle mass loss, and GH deficiency is associated with blood glucose and lipid changes, BMI and waist gain, and decreased BMD.18 All of this may worsen or delay the patient's rehabilitation capacity after TBI.
Factors that promote metabolic change after TBI.
| • Involvement of hypothalamic centres of appetite, satiety or energy homeostasis. |
| • Changes in circadian rhythm, inactivity, or reduced ability to perform aerobic activity (sequelae of TBI). |
| • Need for drugs with effects on weight and metabolism to treat post-TBI complications (corticosteroids, antidepressants, anticonvulsants, etc.). |
| • Hypopituitarism. |
They have been mainly reported with GH deficiency and include higher levels of depression, poorer scores in physical health perception, fatigue and energy levels, emotional well-being, pain, and general health tests.
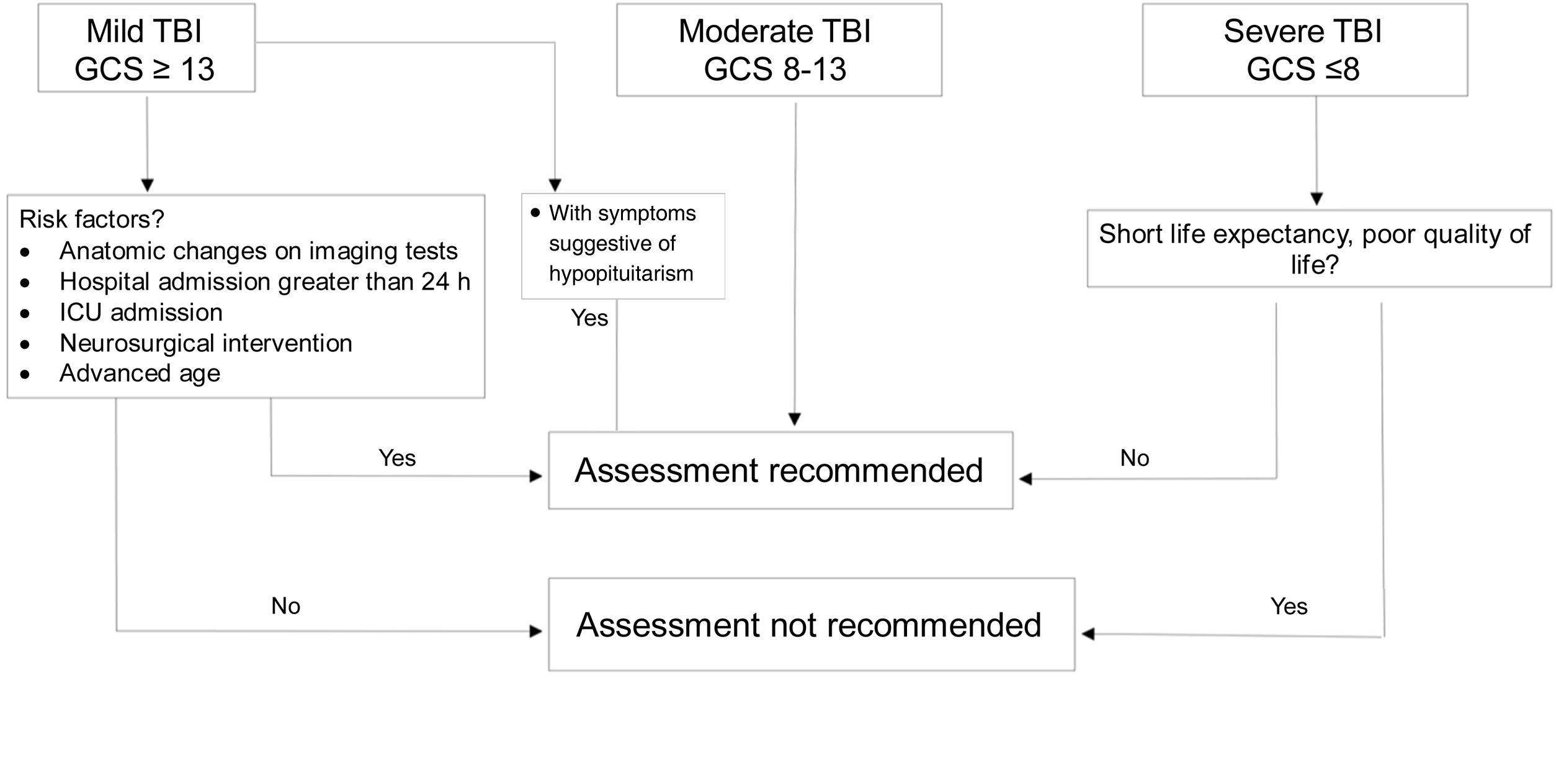
DiagnosisWhich patients should be assessed?If TBI is mild, assessment is not considered cost-effective because prevalence of hypopituitarism in mild cases not requiring admission or subsequent intervention is <1%.20 However, in “complicated” mild cases (hospital admission >24h, neurosurgical intervention, monitoring in critical care unit or anatomical changes in imaging tests),7 in patients with loss of consciousness or episode of post-traumatic amnesia >30min and in those who develop water and electrolyte imbalance or acute adrenal insufficiency, diagnostic assessment is recommended.18,20
Severe cases with sequelae and reduced life expectancy do not benefit from replacement therapy or screening.
In patients with moderate severity, universal screening is recommended (Fig. 2).
When should the assessment be done?
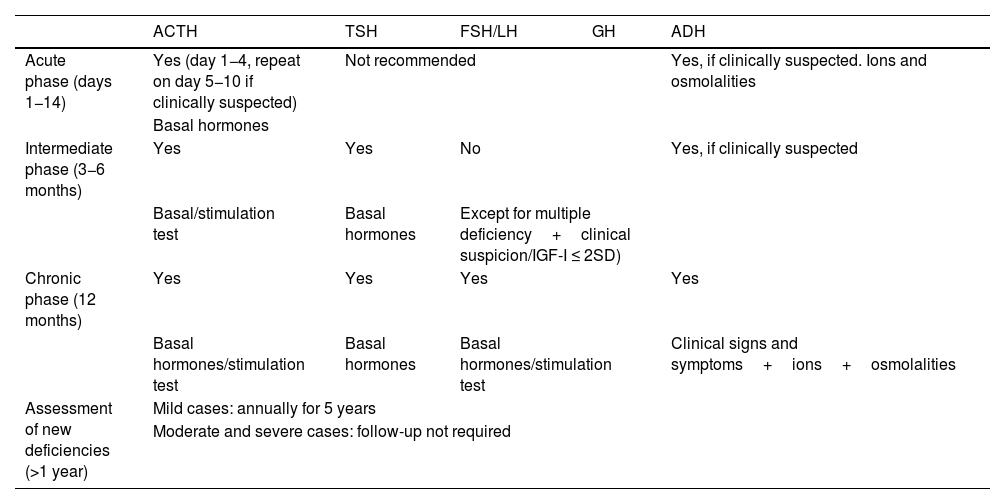
In the acute phase (14 days after TBI), the only hormonal axis to be systematically assessed is the corticotropic axis with basal cortisol measurement between day 1 and 4 after TBI, which will be repeated between days 5−10 if hypoadrenalism is clinically suspected. ADH deficiency should be ruled out if hypernatremia and/or polyuria are present.15 Routine assessment is not recommended for all other axes.
The thyrotropic, gonadotropic and corticotropic axes should be assessed at 3−6 months after TBI. However, GH deficiency must be assessed (together with a complete hormonal study) one year after TBI7 (Table 4). Some selected cases, with multiple hormone deficiencies, clinical signs consistent with somatotropic deficiency, or with IGF-I ≤ 2 SD for age and sex (56), may benefit from early assessment (at 3−6 months).
When should hypopituitarism be assessed after TBI?
| ACTH | TSH | FSH/LH | GH | ADH | |
|---|---|---|---|---|---|
| Acute phase (days 1−14) | Yes (day 1−4, repeat on day 5−10 if clinically suspected) | Not recommended | Yes, if clinically suspected. Ions and osmolalities | ||
| Basal hormones | |||||
| Intermediate phase (3−6 months) | Yes | Yes | No | Yes, if clinically suspected | |
| Basal/stimulation test | Basal hormones | Except for multiple deficiency+clinical suspicion/IGF-I ≤ 2SD) | |||
| Chronic phase (12 months) | Yes | Yes | Yes | Yes | |
| Basal hormones/stimulation test | Basal hormones | Basal hormones/stimulation test | Clinical signs and symptoms+ions+osmolalities | ||
| Assessment of new deficiencies (>1 year) | Mild cases: annually for 5 years | ||||
| Moderate and severe cases: follow-up not required | |||||
Diagnostic methods for hypopituitarism after TBI are the same as for other causes, but test selection or interpretation varies.
Diagnosis of adrenal insufficiency in the acute phase is made by basal cortisol measurement. In general, a cortisol value >15μg/dL excludes ACTH deficiency, <3μg/dL confirms it, and intermediate values require a stimulation test for confirmation. However, in this acute phase, these cut-off points are not applicable, as physiological elevation of cortisol levels occurs. A cut-off point of 11μg/dL (300mmoL/L) has been proposed, below which the diagnosis of adrenal insufficiency would be established and replacement therapy would be required.15,21 A stimulation test in this early setting is not clinically appropriate in most cases. Insulin-induced hypoglycaemia is inappropriate for safety reasons, and the ACTH stimulation test would not discriminate acute cases in which failure had not yet been established. In later phases, the ACTH stimulation test with low doses (1−2μg) is preferable to avoid supraphysiological stimuli that could modify the results. The new competitive immunoassays showed greater sensitivity and specificity in the detection of adrenal insufficiency, with lower cortisol cut-off points. Cortisol levels after stimulation >18μg/dL (500nmol/L) according to standard immunoassays or >13.4μg/dL (374nmol/L) according to new immunoassays, exclude deficiency.
Assessment of hypothyroidism includes basal free T4 and TSH measurements. The TRH stimulation test did not show greater diagnostic precision.18
For hypogonadism, basal gonadotropin measurements with sex steroids and menstrual history in premenopausal women are sufficient to establish the diagnosis.
With regard to GH deficiency, IGF-I levels are usually a reliable marker of deficiency. In the presence of multiple pituitary deficiencies, decreased IGF-I will be sufficient for diagnosis.18 All other cases require confirmatory testing, such as insulin-induced hypoglycaemia, glucagon testing, or stimulation with GHRH and arginine or other GH secretagogue as GHRH+GHRP-6.
In isolated deficiencies, this will be confirmed with a second test. Insulin-induced hypoglycaemia in TBI is less useful because it is contraindicated due to the risk of convulsion after TBI, and also has a lower sensitivity in this setting. GH measurement after stimulation with macimorelin (GHRYVELIN®) has been shown to be useful because its diagnostic accuracy is not influenced by factors that modify GH secretion, such as obesity, sex, or age.22 If other hormone deficiencies are present, they should be adequately replaced when assessing GH reserve.
ADH deficiency should be suspected in the presence of polyuria (diuresis >40mL/kg/d). Increased plasma osmolality with a urinary/plasma osmolality ratio <2 supports the diagnosis. Measurement of basal and stimulated copeptin may be useful for diagnosis after the acute phase of TBI.
With regard to follow-up, recovery and appearance of new deficiencies has been detected 3 and 5 years after injury, particularly in mild cases, so annual clinical and biochemical monitoring is recommended for at least 5 years to detect both possible recoveries and new deficiencies.23 In moderate and severe cases, due to the low probability of recovery of hormone deficiencies, replacement therapy should be monitored. If no hormone deficiencies are detected after 12 months, subsequent monitoring is not required, unless clinically suspected (Table 4).
TreatmentIt involves replacement of deficient hormonal axes as in other cases of hypopituitarism and differs in the acute and chronic phase after TBI.
In the acute phase, adrenal axis replacement should be performed after confirmation of deficiency while there is insufficient evidence to recommend replacement of the thyroid, somatotropic, and gonadotropic axis.7 It is recommended to treat hypocortisolism with hydrocortisone (oral 30mg/day in stable patients and intravenous 50−150mg/8h or continuous infusion at 15mg/h in critically ill and unstable patients). The dose will be adjusted according to clinical response and will be maintained at stress doses while the patient requires vasopressor treatment. Subsequently, it is recommended to continue with physiological doses (15−20mg/d) until reassessment of the axis can be performed. Routine treatment with corticosteroids in critically ill patients after TBI is not indicated as benefits have not been proven. GH plays a crucial role in cell proliferation and neuronal repair mechanisms, but there are no clinical studies assessing the effects of GH replacement therapy in the acute phase of TBI, and it is therefore not recommended. There is also no evidence to recommend thyrotropic and gonadal replacement therapy. Treatment with thyroid hormone in critically ill patients for conditions other than TBI has not shown improvement.
In contrast, in the chronic phase and with established hormone deficiencies, treatment is recommended for pituitary hormone deficiencies of any etiology. GH replacement therapy improved cognitive function, including memory, information processing speed, vocabulary, executive function, or verbal learning and is considered as a key treatment associated with rehabilitation because it has been shown to increase muscle strength, aerobic capacity, and improve body composition after one year of treatment,24 although there are no studies in TBI. Quality of life impairment appears to be greater in cases of TBI as compared to other causes despite apparently milder biochemical deficiencies, but they achieve greater benefit with treatment maintained in the long term. Replacement of all other axes was not specifically assessed in TBI and should follow the general indications for hypopituitarism.
ConclusionsHypopituitarism secondary to TBI is common and leads to increased morbidity, as well as a subsequent limitation in functional capacity and recovery. Aside from the direct damage caused by TBI, complex mechanisms of excitotoxicity, ischaemia, inflammation, or immunity have been identified that would explain the occurrence of hypopituitarism. Diagnosis requires homogeneous screening criteria, patient selection, timing, and diagnostic methods. In the acute phase, detecting and treating hypocortisolism is vital, while all other axes do not benefit from early interventions. In chronic stages, hypopituitarism, in addition to classical manifestations, is associated with neurocognitive impairment and poorer quality of life. Treatment of hormone deficiencies in these cases has been shown to improve quality of life, metabolic changes, and body composition, improving the prognosis of these patients.
Authors’ contributionAll authors contributed to the writing of the paper, critically reviewed its content and approved the final version.
AcknowledgementsThis document has been reviewed by an external scientific committee and has been approved by the SEEN Board of Directors. The authors would like to thank the reviewers appointed by the Sociedad Española de Endocrinología y Nutrición for their suggestions and corrections, which have contributed to the improvement of the document.
FundingThis article has not received any funding.
Conflict of interestNone.
Link to full document on SEEN page https://www.seen.es/portal/areas-conocimiento/neuroendocrinologia/documentos/consensos-guias/hipopituitarismo-traumatismo-craneoencefalico-adulto.
