




Erdheim–Chester disease (ECD) is a rare form of non-Langerhans' cell histiocytosis first reported in 1930. This disease of unknown aetiology manifests in adults around their 40s–60s, predominantly in males. It is a multisystemic disease featuring bone involvement in all cases, with a predilection for the long bones.1
Some 50% of cases feature extraskeletal clinical signs with central nervous system involvement, including central diabetes insipidus and orbital, cardiac, pulmonary, pleural, vascular (great vessels), retroperitoneal and cutaneous involvement. The prognosis depends on the extent of organ involvement; cardiovascular and central nervous system involvement are associated with a more serious disease course.2
We report a case detected in an endocrinology practice in which ECD was suspected given the existing association between diabetes insipidus and bone involvement (symmetric osteosclerosis of the long bones) previously reported by the patient.3 Imaging tests and, in particular, bone biopsy were key to arriving at the diagnosis.
The patient was a 50-year-old man in follow-up by rheumatology for migratory arthralgia with bone scintigraphy showing diffuse radioactive tracer deposition in the long bones of the arms and legs, consistent with osteosclerosis. At an appointment in early 2019, the patient reported that he drank around nine litres of water per day due to intense thirst, whereupon he was referred for assessment by endocrinology. The possibility of polydipsia was considered, but ruled out following a dehydration test, with results consistent with central diabetes insipidus and an excellent response to desmopressin.
The test results were:
- -
Hormone tests: no other pituitary or gonadal deficiencies.
- -
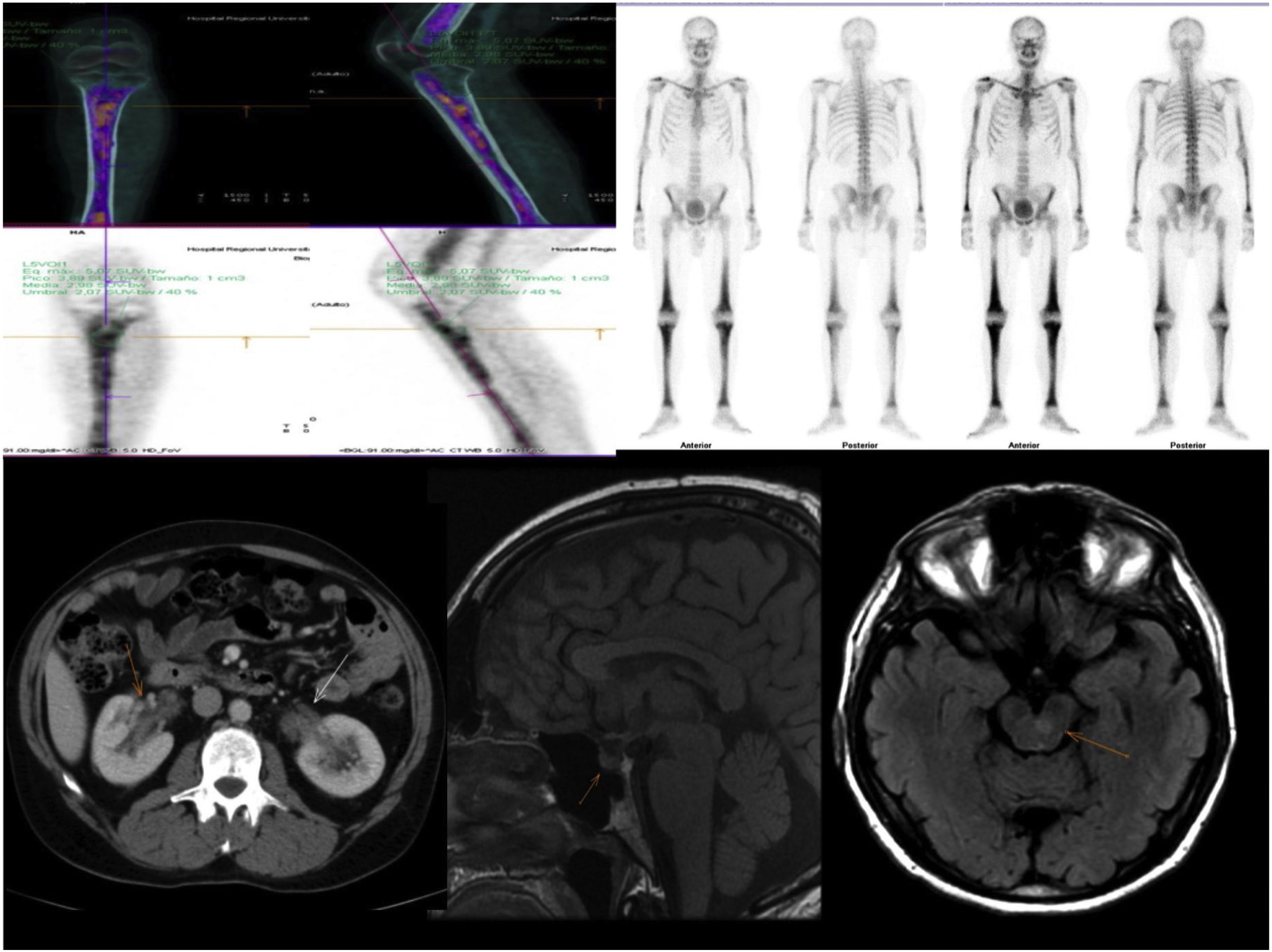
Imaging tests: (Fig. 1).
 Figure 1.
Figure 1.Radiological findings: Characteristic bone involvement on PET/CT and bone scintigraphy, CT of the abdomen with no pathological findings in the kidney; MRI of the brain. Sagittal slices in T1 and axial slices enhanced in T2 FLAIR. The sellar region shows no significant pathological abnormalities. Multiple hyperintense signal abnormalities are identified on T2 FLAIR in the brain stem, especially in the area of the pons.
(0.33MB). - -
Computed tomography (CT) of the chest, abdomen and pelvis: lung parenchyma featuring a ground-glass pattern with interstitial thickening in the right lower lobe consistent with an inflammatory process, without any other findings of note.
- -
Magnetic resonance imaging (MRI) of the brain: multiple small signal abnormalities on both sides of the midline of the brain stem and cerebellum. No pathological abnormalities were seen in the pituitary gland.
- -
Positron emission tomography/computed tomography (PET/CT): diffuse involvement of bone marrow of long bones, with greater uptake in the left proximal tibial metaphysis. Hypermetabolic focus in left pons in relation to active lesion. Increases in hypermetabolic density in right lung consistent with inflammatory signs which, along with the hypermetabolic lymphadenopathy reported in the right paratracheal and subcarinal region, were suggestive of an inflammatory process.
- -
Bone biopsy of proximal tibial metaphysis: lesions suggestive of histiocytosis. CD68+, CD1a and S100 negative. Positive for BRAF V600E mutation.

With the above findings, the patient was diagnosed with ECD. In this case, the patient was referred to haematology to start treatment as mandated by the central nervous system involvement he exhibited.
In adults, the most common type of non-endocrine involvement in ECD is bone involvement, in the form of metadiaphyseal osteosclerosis of the long bones (80%–95% of patients), which can cause bone pain or be asymptomatic.4
Regarding endocrine manifestations,5diabetes insipidus develops in a third of patients, often as the first sign.6,7 Anterior hypopituitarism is found in the vast majority of patients when complete pituitary function is evaluated. The following may appear, in order of frequency: growth hormone (GH) deficiency, hyperprolactinaemia, gonadotropin deficiency,8 thyrotropin deficiency and corticotropin deficiency. On MRI, infiltration of the pituitary gland with pituitary stalk thickening is found in 24% of patients.
Testicular failure appears in half of patients, with bilateral testicular infiltration on ultrasound. Adrenal infiltration on CT is seen in a third of patients and may manifest as adrenal insufficiency.9
Central nervous system involvement occurs in around 30%–50% of cases. It sometimes leads to severe disability and constitutes the main factor in a poor prognosis. Pyramidal and cerebellar syndromes are the earliest and most common neurological signs.
One out of every four patients develops exophthalmos due to infiltration of retro-orbital tissue.
The most common cardiovascular sign is the coated aorta sign. A wall mass due to infiltration—the so-called pseudotumour of the right atrium—may be observed. It causes infiltration of the pericardium and coronary arteries with myocardial infarction.
In the retroperitoneal area, abdominal CT reveals infiltration of perirenal fat, with characteristic imaging of hairy kidneys in 60% of patients. One third have retroperitoneal fibrosis.
Skin manifestations consist of xanthelasma, while lung involvement is seen in half of cases.2,4
Concerning molecular pathogenesis, ECD is a clonal neoplastic disorder driven by mutations in the mitogen-activated protein kinase (MAPK) pathway (RAS/RAF/MEK/ERK), which are targets of molecular therapy in histiocytosis.
It is associated with myeloid leukaemia, myelodysplastic syndrome, other systemic histiocytoses and other autoimmune diseases.1
Arrival at a diagnosis requires joint analysis of histopathological characteristics and clinical, radiological and molecular findings.
Biopsy of affected tissues is required in all cases, not only to confirm the diagnosis with identification of histiocytes featuring a characteristic immunohistochemistry pattern, but also to enable identification of associated mutations for treatment purposes.
The most significant histopathological findings consist of infiltration by “foamy” histiocytes. ECD can be distinguished from Langerhans' cell histiocytosis by the immunohistological characteristics of its histiocytes, which are positive for CD68 and negative for CD1a and CD207 (which are characteristic of Langerhans' cell histiocytosis). Once the histological diagnosis is confirmed, the BRAF V600E mutation is verified in all patients. If negative, abnormalities in other MAPK pathway genes should be investigated.
Radiological findings such as osteosclerosis, infiltration of perirenal fat and proliferation of soft tissue surrounding the aorta are highly characteristic. PET/CT scans show pathological uptake that is useful for establishing the diagnosis, guiding biopsies and assessing extent and treatment response.
Treatment decisions are difficult to generalise and should be adapted to the characteristics of each individual patient and the seriousness, location and extent of their lesions. An expert consensus was published in 2014 (Diamond et al.)10 and updated in 2020 (Goyal et al.).11 Most patients will require treatment, except those who have asymptomatic forms with involvement of a single non-vital organ (bone). Conventional treatments are available, such as interferon, with extensive experience in its use, as well as interleukin-1 and interleukin-6 receptor antagonists. Targeted treatments are also available, with BRAF inhibitors such as vemurafenib, MEK inhibitors such as cobimetinib or a combination of the two. Other available treatments are mTOR inhibitors such as sirolimus and tyrosine kinase inhibitors.
At present, BRAF inhibitors are first-line drugs. For BRAF V600 mutation-negative patients, it is advisable to look for other MAPK pathway abnormalities, which can be treated with a MEK inhibitor.
In conclusion, the first sign of central nervous system involvement due to ECD in our patient was diabetes insipidus.12 The manifestation of prior skeletal signs as well as characteristic radiological and histological findings led to the patient's definitive diagnosis; hence, a comprehensive medical history by endocrinology was of particular importance in suspecting this rare disease.
Please cite this article as: Vallejo Herrera MJ, Sánchez Torralvo FJ, Vallejo Herrera V, Olveira Fuster G, Pérez de Pedro I. Enfermedad de Erdheim-Chester: diagnostico en endocrinología. Endocrinol Diabetes Nutr. 2022;69:444–446.
