





Primary bilateral macronodular adrenal hyperplasia (PBMAH) accounts for <2% of cases of Cushing's syndrome. The majority of patients present with no obvious steroid excess it means with autonomous cortisol secretion (ACS). The classic treatment for patients with overt Cushing's syndrome is bilateral adrenalectomy, but unilateral resection of the larger adrenal gland can result in clinical and/or biochemical remission in >90% of cases, especially in cases of ACS. In this article, a series of 32 cases with PBMAH is described. Most of the cases of PBMAH had ACS, except for one case with overt Cushing's syndrome. A study of aberrant receptors was performed in six patients, being negative in three cases, positive in the metoclopramide test in two cases and positive in the metoclopramide test and in the mixed meal test in another patient. The patient with overt Cushing's syndrome was treated with adrenostatic therapy achieving biochemical control, while two patients with ACS underwent unilateral adrenalectomy with resection of the largest adrenal gland, demonstrating hypercortisolism remission and improvement of cardiovascular risk factors after surgery. This article describes a series of 32 cases of PBMAH and offers a comprehensive review of PBMAH.
La hiperplasia suprarrenal macronodular bilateral primaria (HSMBP) representa <2% de los casos de síndrome de Cushing. La mayoría de los pacientes se presentan sin exceso evidente de secreción de esteroides, es decir con secreción autónoma de cortisol (SAC). El tratamiento clásico para pacientes con síndrome de Cushing florido es la suprarrenalectomía bilateral, pero la resección unilateral de la glándula suprarrenal más grande puede resultar en remisión clínica o bioquímica en>90% de los casos, especialmente en los casos de SAC. En este artículo se describe una serie de 32 casos con HSMBP. La mayoría de los casos tenían SAC, excepto un caso con síndrome de Cushing florido. Se realizó estudio de receptores aberrantes en 6 pacientes, que fue negativo en 3 casos, positivo en el test de metoclopramida en 2 y positivo en el test de metoclopramida y en el test de comida mixta en otro paciente. El paciente con síndrome de Cushing florido fue tratado con terapia adrenostática y alcanzó un adecuado control bioquímico, mientras que a 2 pacientes con SAC se les hizo adrenalectomía unilateral con resección de la glándula suprarrenal más grande y se evidenció remisión del hipercortisolismo y mejoría de los factores de riesgo cardiovascular tras de la cirugía. Este artículo describe una serie de 32 casos de HSMBP y ofrece una revisión exhaustiva sobre el tema.
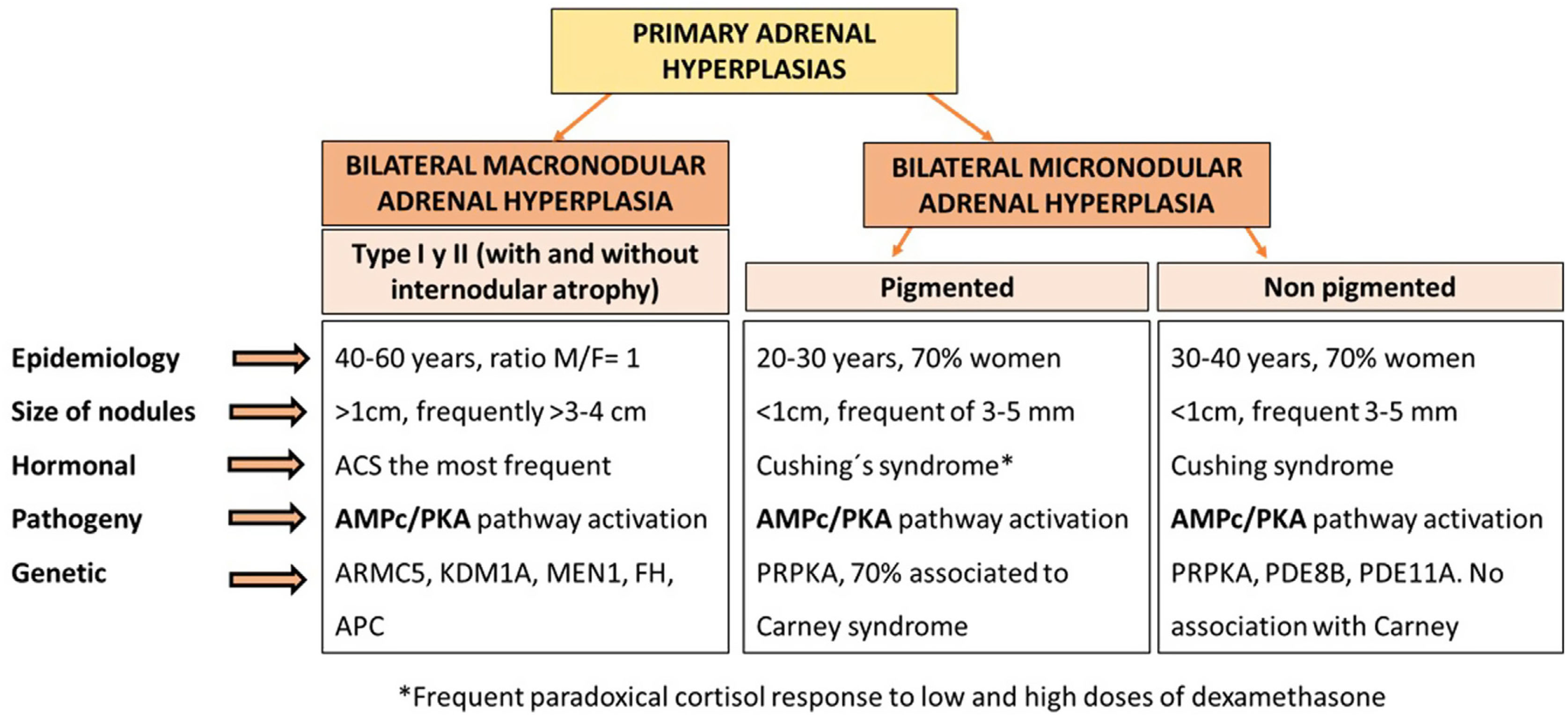
Primary adrenal aetiologies account for 15–20% of the causes of endogenous Cushing's syndrome. The most common cause is unilateral adrenocortical cortisol producing adenoma, while bilateral adrenal hyperplasia is responsible for only 10% of cases of adrenal Cushing's syndrome.1 Primary adrenal hyperplasia includes cases of primary bilateral macronodular adrenal hyperplasia (PBMAH) and primary bilateral micronodular adrenal hyperplasia. Both conditions generally causes low or suppressed levels of adrenocorticotropic hormone (ACTH), but differ characteristically in terms of genetic, clinical, hormonal, and radiological presentation2 (Fig. 1).

The new World Health Organization (WHO 2022) classification of adrenal cortical proliferations reflects translational advances in the fields of endocrine pathology, oncology and molecular biology and recognises the importance of structural and functional correlations.3 In this new classification, the causes of cortical nodular adrenal disease include (a) sporadic nodular adrenocortical disease, (b) bilateral micronodular adrenal cortical disease, and (c) bilateral macronodular adrenal cortical disease (corresponding to primary bilateral macronodular adrenal hyperplasia in this review). This classification highlights the importance the frequent genetic origin of PBMAH and suggests that the term hyperplasia is not appropriate due to the characteristic clonal and/or neoplastic pattern that the disease presents. However, the use of these terms is not yet widespread in clinical practice.
In this article, a series of 32 cases with PBMAH is described, focusing on the specific treatment of hypercortisolism in these patients. In addition, it contains a comprehensive review of the most important aspects of the epidemiology, diagnosis, genetics, pathophysiology and treatment of PMBAH, highlighting the latest developments in this field in the last years.
Case series of patients with PBMAHA total of 32 cases with PBMAH out of the 732 cases of adrenal incidentalomas were identified in the Ramón y Cajal Hospital (Madrid, Spain) in the period between 2013 and 2019. The mean age was 64.2±11.2 years and 59.4% were females. Only one patient had overt Cushing's syndrome, while the remaining cases presented with ACS (possible ACS (dexamethasone suppression test (DST)>1.8μg/dl), in 21 patients, and confirmed ACS (DST>5μg/dl), in 10 patients. The mean levels of cortisol post-DST were 5.7±3.97μg/dl and the mean tumour size of adrenal nodules was 30.2±12.96mm. In all, 84.4% of the patients had one or more comorbidities potentially attributable to hypercortisolism, with hypertension (71.9%), dyslipidaemia (56.3%) and type 2 diabetes (43.8%) being the most common. Moreover, seven patients had cardiovascular disease and another seven had obesity. Mean glucose levels were 111.2±39.0mg/dl and mean HbA1c was 7.1±2.12% (results available only in 15 patients). A genetic study for ARMC5 mutations was performed in the patient with overt Cushing's syndrome, but results were still pending. Investigation of aberrant receptor expression (following the Lacroix protocol, as we have previously described4) was performed in six patients. It was negative in three patients (one of the patients with overt Cushing's syndrome), positive in two patients in the metoclopramide test (cortisol increase of 63% in one and of 50% in the other patient), and positive in another patient in both the metoclopramide and the mixed meal test (cortisol increase of 48% and 62%, respectively). The patient with the positive mixed meal test was treated with 120μg/month of lanreotide with no response, and one of the patients with a positive metoclopramide test received amitriptyline with no response either. Two patients with ACS underwent unilateral adrenalectomy with the resection of the largest adrenal gland. A significant improvement of cardiovascular risk factors was reported after surgery. The patient with overt Cushing's syndrome was treated with ketoconazole (median doses of 1000mg/day at the last visit), and after a follow-up of four years, hypercortisolism remained properly controlled. As in other PBMAH series,5 in our study we found that ACS was the most common presentation of PBMAH, and these patients have a high prevalence of cardiometabolic comorbidities. Although in our series only 50% of the patients who underwent a study of aberrant receptors had a positive result in the study, the lower percentage in comparation with other studies6,7 may be related to the low number of patients tested in our cohort. However, in accordance with what has been previously described, we observed that response to specific medical treatment in these cases is usually ineffective, or only a transient response is observed.8,9 Nevertheless, unilateral adrenalectomy with resection of the largest adrenal gland is usually an effective treatment to control hypercortisolism and reduce cardiometabolic risk in patients with PBMAH, especially in those with ACS.10 Another important point is the recommendation to perform a genetic study in patients with PBMAH. Although in our series only the patient with overt Cushing's syndrome underwent genetic testing (for unknown reasons), the current recommendation is to perform a genetic study in all patients with PBMAH, since germline pathogenic variants in ARMC5 are found in 15% of patients with apparently sporadic PBMAH and 80% of patients with evident familial presentation.11–13 Furthermore, KDM1A inactivation explains about 90–100% of food-dependent Cushing's syndrome.14,15 The identification of these mutations in the family may lead to an earlier diagnosis of hypercortisolism and optimisation of its management.
Epidemiology and definition of PBMAHPBMAH is characterised by bilateral benign adrenal macronodules larger than 1cm potentially responsible for variable levels of cortisol excess. It is considered a rare disease, representing less than 2% of cases of Cushing's syndrome. Overt Cushing's syndrome is present in a minority of cases. Most frequently, PBMAH is identified incidentally by imaging performed for an unrelated reason. A significant percentage of the latter patients have ACS.5 In this situation, hypercortisolism usually has an insidious course, progressing slowly over several years, which in part explains why it is a usually underdiagnosed condition. In this context, in one of our recent studies, we found that up to 30% of patients with bilateral adrenal disease and associated hypercortisolism and 4% of all adrenal incidentalomas met criteria for PBMAH.4
PBMAH is usually defined by the existence of adrenal enlargement with multiple bilateral benign adrenocortical nodules larger than 1cm that can cause cortisol excess independently of ACTH from the pituitary. The size of the nodules is one of the key factors for the differentiation between macronodular and micronodular forms of adrenal hyperplasia, since in micronodular forms the adrenal nodules are generally smaller than 1cm16 (Fig. 1). Two histological forms of PBMAH have been differentiated, based on whether or not there is cortical internodular atrophy: type I PBMAH, which is characterised by internodular atrophic tissue, and the more common type II PBMAH, which exhibits diffuse hyperplasia and absence of normal or atrophic internodular tissue.5 Nevertheless, despite this definition, adrenal hyperplasia is a highly heterogeneous condition, since the size of the adrenal nodules can variable widely, from 1cm to more than 5–10cm. Also, the degree of hypercortisolism is variable, from non-functional to ACS and overt Cushing's syndrome.17 Asymptomatic cases without associated hypercortisolism have been observed in relatives carrying mutations in ARMC5, detected in the genetic study due to a family history of PBMAH.18 Moreover, it must be taken into account that PBMAH can present with asynchronous nodular hyperplasia, initially in one gland and years later in the contralateral adrenal gland.19
Sporadic PBMAH predominantly affects women, with a female to male ratio of 2:3, while in patients with mutations in ARMC5, it affects men and women equally.2 The average age of diagnosis is 45–60 years, and in patients with McCune-Albright syndrome, PBMAH is usually diagnosed at 5–10 years of age.20
PathophysiologyOne of the best-known mechanisms involved in the pathogenesis of PBMAH is the abnormal regulation of the adrenal cortex by aberrant G-protein coupled receptors that leads to activation of the AMPc/PKA pathway, with the consequent abnormal stimulation of cortisol biosynthesis, and, to a lesser degree, adrenal enlargement. It is estimated that approximately 75–80% of patients with PBMAH have aberrant receptors that are stimulated by ligands different from ACTH.6,7
There are some receptors with ectopic expression such as gastrointestinal inhibitory peptide (GIP), catecholamines, vasopressin type 2 and 3 (V2–V3), serotonin type 7 (5-HT7), angiotensin II type 1 (AT1); and other receptors with eutopic expression but overexpressed such as vasopressin type 1 (V1), LH/hCG, serotonin type 4 (5-HT4) and leptin. In patients with GIP receptors, food intake induces an increase in cortisol levels, with normal concentrations during fasting periods; those patients with V1 vasopressin receptors have increased cortisol with postural changes; those cases with beta-adrenergic receptor expression have cortisol elevations in parallel to the endogenous increases in catecholamines (in response to orthostasis, hypoglycaemia, or exercise) and those with LH/hCG receptors can develop hypercortisolism with pregnancy or menopause.21 Up to 50% of patients have a positive response to four or more stimuli, with vasopressin (V1) and serotonin (5-HT4) responses being the most common.6,7,21 However, it should be noted that although PBMAH is the best-known condition in which aberrant receptors are involved, there are other situations in which they may be present. In summary, 20–30% of unilateral or bilateral adrenal incidentalomas with ACS have aberrant receptors, and their presence is closely linked to hypercortisolism, but not other forms of steroid excess.22,23
Moreover, the intra-adrenal production of ACTH by the steroidogenic cells is another mechanism that has been involved in the pathogenesis of PBMAH. Aberrant receptors that regulate local ACTH secretion through a paracrine/autocrine mechanism that would stimulate glucocorticoid production and adrenal nodular growth have been detected. In fact, following the discovery of this mechanism by Lousiet et al. in 2013,24 the term ACTH-independent macronodular adrenal hyperplasia is no longer used. In Lousiet's research, proopiomelanocortin (POMC) mRNA was detected in 26 PBMAH samples tested, moderate/strong immunostaining for ACTH was detected in all except for one sample, and there was no ACTH staining in normal adrenal cortex or cortisol-secreting adenomas. In addition, ACTH secretion was confirmed by the presence of an ACTH gradient on adrenal vein sampling in two patients with PBMAH.24
Other mechanism such as the activation of wnt/b-catenin pathway have been implicated in the stimulation of adrenal node growth.25
GeneticsThe genetics of PBMAH is one of the fields, in which there has been major advances in the last years, identifying almost 10 genes involved in its pathogenesis, with mutations in ARMC5 being the most important.26
It is estimated that approximately 5% of PBMAH cases are part of familial syndromes such as McCune-Albright syndrome, multiple endocrine neoplasia syndrome type 1 (MEN1), familial adenomatous polyposis (FAP), or hereditary leiomyomatosis and renal cell cancer syndrome.27–29 Adrenal involvement has been described in 20–40% of patients with MEN1. However, endoscopic ultrasound detected adrenal lesions in up to 73% of the patients.30 Adrenal lesions usually develop in patients with mutations in exons 2 and 10.31 However, overall, PBMAH cases are anecdotal in MEN1, with only 17 cases having been described in the French series of 715 patients with MEN1.32 The prevalence of adrenal lesions in patients with FAP is 13%. Most adrenal masses in these patients are nonfunctioning and benign adrenal lesions.33 McCune-Albright syndrome is a genetic syndrome characterised by fibrous dysplasia of bone, hyperpigmentation of the skin with café-au-lait spots, and GnRH-independent precocious puberty.34,35 It is the familial syndrome most frequently associated with PBMAH, with up to 8% of patients affected by PBMAH.36 However, by far the most frequent cause of hereditary PBMAH are germline mutations in ARMC5, present in up to 80% of hereditary cases according to some series37 (Table 1).
Genes associated with PBMAH and adrenal involvement.
| Gene | Clinical manifestations | Adrenal involvement | Bilateral adrenal tumours | PBMAH cases |
|---|---|---|---|---|
| Syndromic cases | ||||
| MEN1, 11q13 (tumour suppressor gene)32 | Primary hyperparathyroidism, pancreatic NETs, pituitary adenomas and adrenocortical tumours | 25–30% | 12.5% | 17 cases described |
| APC, 5q21 (tumour suppressor gene)33 | Multiple colorectal polyps more predisposed to develop colorectal cancer, thyroid tumours and adrenocortical tumours | 16% | 23% | <1% |
| GNAS1, 20q13 (oncogene)35 | Fibrous bone dysplasia, café-au-lait spots, skin hyperpigmentation and peripheral precocious puberty | 8–10% | >50% | 8% |
| FH, 1q42.1 (tumour suppressor gene)29 | Leiomyomas and increased risk of kidney cancer | 7.8% | 15% | <1% |
| Non-syndromic cases | ||||
| ARMC5, 16p11.2 (tumour suppressor gene) | Possible association with intracranial meningioma: Lee 2005,40 Alencar 2014,37 Elbet 2015,41 Jojima 2020,42 Ferreira 202043 | >90% | >90% | >90% from the age of 40. More severe cases of hypercortisolism vs. ARMC5 wild type |
| KDM1A, 1p36.12 (tumour suppressor gene) | Possible association with myeloma and monoclonal gammopathy of uncertain significance | >90% | >90% | >90% from the age of 40. Associated with food-dependent Cushing's syndrome |
ARMC5: armadillo repeat containing 5; APC: adenomatous polyposis coli; FH: fumarate hydratase; PBMAH: primary bilateral macronodular adrenal hyperplasia; MEN1: multiple endocrine neoplasia syndrome type 1; KDM1A: lysine demethylase 1A; NET: neuroendocrine tumour.
Germline mutations in ARMC5 reduce the ability of cells to secrete steroids, which is consistent with previously reported ineffective steroidogenesis in PBMAH patients, resulting in lower urinary free cortisol (UFC) levels than expected for the size of the adrenals and an increase in cortisol precursors. ARMC5 is located in chromosome 16p11.2 and behaves as a tumour suppressor gene, causing apoptosis when transfected into H295 adrenal carcinoma cell lines.11 ARMC5 inactivation decreases the expression of the ACTH receptor (MC2R) and steroidogenic enzymes.11 Currently, a total of 119 genetic pathogenic variants of ARMC5 have been identified in patients with PBMAH, both at the germline and at the somatic level.38 It is estimated that 10–55% of PBMAH patients have germline mutations in ARMC5, with up to 80% in familial cases (autosomal dominant inheritance, variable clinical expression), and around 15% in apparently sporadic cases.13 ARMC5 mutations are more frequent in cases with overt Cushing's syndrome, ranging from 28% to 55%, than in cases of ACS in which the prevalence is about 10%.2 The presence of the mutation is also more common in those patients with larger adrenal glands and with multiple macronodules.13,39 Its presence has been associated with a higher frequency of aberrant beta-adrenergic and vasopressin V1 receptors. Nonetheless, it has not been associated with response to GIP, unlike mutations in KDM1A.11 In fact, mutations in ARMC5 and KDM1A have recently been found to be exclusive molecular events14,38 (Table 1).
In relation to the germline mutations in ARMC5, it has been suggested that it could play a pathogenic role in the development of intracranial meningiomas. The coexistence of PBMAH and intracranial meningioma was first described in 2005 in two patients in whom both conditions coexisted.40 Subsequently, several cases have been described.37,41–43 For this reason, some authors recommend performing brain MRI to rule out meningioma in patients carrying the mutation36 (Table 1).
One of the most recent discoveries in PBMAH is the role of mutations in KDM1A in GIP-mediated cases of Cushing's syndrome. In the study led by Chasseloup,15 of 17 patients with PBMAH and GIP-responsive Cushing's syndrome, 100% of patients had a germline mutation in KDM1A, compared with 0% of the patients in the control group. In addition, they found that carriers of this mutation had a higher risk of developing monoclonal gammapathies, so they recommend that serum protein electrophoresis should be mandatory in patients with this mutation. Another more recent study found that 90% of cases of food-dependent Cushing's syndrome carried inactivating mutations in KDM1A.14 Cortisol secretion in these patients is stimulated by food intake and the diagnosis can be made by analysing the circadian rhythm of cortisol levels, with low fasting cortisol and elevated cortisol secretion after a meal or an oral glucose load. PBMAH cases with mutations in KDM1A also showed overexpression of other aberrant receptors, such as LH/hCG, angiotensin II receptor type 1, and kisspeptin receptor (KISS1R).15 Unlike ARMC5 mutations, which affect men and women equally, KDM1A mutations are more common in women14,15 (Table 1). The discovery of genetic alterations such as ARMC5 and KDM1A in PBMAH allows early detection of PBMAH in relatives, which has the potential to reduce the consequences of long-term cortisol hypersecretion as a result of earlier diagnosis.38 Despite numerous pan-genomic sequencing studies, to date, neither ARMC5 nor KDM1A pathogenic variants have been described in unilateral sporadic benign lesions or malignant adrenocortical tumours.38
Other mutations have been described, such as mutations in MCR2,44 protein regulatory subunit kinase A (PRKACA),45 phosphodiesterase 11A (PDE11A)46 and 8B (PDE8B),47 but these mutations are much less frequent.28
Biochemical diagnosisThe biochemical diagnosis will depend on whether the patient has a clinical suspicion of Cushing's syndrome, in which case we will carry out confirmatory biochemical studies aimed at hypercortisolism,48 or if the patient has bilateral adrenal incidentalomas. In this second case, screening for pheochromocytoma, hypercortisolism and primary aldosteronism (in the presence of hypertension and/or hypokalaemia) should be performed.49,50 The recommended test for initial screening for hypercortisolism in adrenal incidentalomas is the 1-mg dexamethasone suppression test (DST). On the other hand, although it is anecdotal, cases of co-secretion of aldosterone, oestrogens and androgens have been described.19,51 In cases with abnormal results in the DST (the most sensitive cut-off for cortisol post-DST for the identification of patients with ACS has been defined at 50 nmol/l [1.8μg/dl]49), other studies should be considered to assess the degree of hypercortisolism by determining UFC and serum or nocturnal salivary cortisol.52 It should be considered that since cortisol secretion may be food dependent, fasting morning studies may lead to false-negative test results. ACTH-dependence must be ruled out by measuring plasma ACTH. However, ACTH is not always fully suppressed, especially in cases with mild hypercortisolism such as ACS.53 Furthermore, the ectopic production of ACTH by the adrenal glands may also explain the existence of unsuppressed levels of ACTH.24 It is estimated that 20% of patients with PBMAH have normal ACTH levels.13 Plasma ACTH levels become progressively suppressed as cortisol levels gradually rise.
Patients with PBMAH have a profile of ineffective steroidogenesis that resembles that of adrenocortical carcinoma, with elevated DHEA-S and other steroid precursors. In this sense, a recent study reported that, based on the study of the steroid profile measured by liquid chromatography coupled to mass spectrometry (LC-MS), cases of PBMAH could be correctly differentiated from other causes of adrenal Cushing's syndrome in 95.2% and differentiate PBMAH cases with and without ARMC5 mutations in 91.7% of subjects.54 It is also important to keep in mind that these patients may present elevations in 17OH-progesterone levels, which may respond to ACTH stimulation. The key parameter to differentiate this condition from congenital adrenal hyperplasia is that in PBMAH there is associated hypercortisolism and usually low ACTH levels.2
Study of aberrant receptorsRegarding the study of aberrant receptors, there are several controversial points in this respect. First, not all authors agree on its usefulness. Those who are in favour of performing the study justify it by its potential to identify candidates for specific treatment with drugs that block these receptors,19,55 while those who are against it indicate that a response to specific long-term treatments is infrequent and that the studies are complex, bothersome for the patient and costly to perform. In addition, more than one aberrant receptor is usually identified in each patient, making it difficult to select a specific receptor for blockade.37 On the other hand, there is no consensus on the type of protocol that is most appropriate. One of the most widely used is the Lacroix protocol, in which serial measurements of ACTH, cortisol, aldosterone, free testosterone, DHEA-S and estradiol are performed every 30–60min for 2–3h after several tests.56 More recently, St-Jean57 recommended performing the studies under suppression with 1mg of dexamethasone every six hours, starting 48h before the tests to avoid the effect of ACTH on steroidogenesis in patients with ACS. In this regard, a previous study of 33 patients with bilateral adrenal hyperplasia found that a significant number of tests would have been erroneously classified as positive or partial responses if dexamethasone suppression had not previously been performed23 (Fig. 2).

Lacroix protocol for the study of aberrant receptors in adrenal disease.53 ACTH: adrenocorticotropic hormone; GIP: gastrointestinal inhibitory peptide; ACS: autonomous cortisol secretion.
Regarding its interpretation, in the case of following the Lacroix protocol, the parameter used to evaluate the response in the different tests is the dynamics of cortisol after the different stimuli. A change of less than 25% in cortisol level is considered unresponsive, a change of 25–49% as a partial response, and a change of 50% or more as a positive response to the test. However, when comparing data from studies performed in vitro with those performed in vivo, some discrepancies are found, suggesting that the criterion of 25–50% change in serum cortisol to establish partial/complete responsiveness should be reassessed, at least when the receptors tested are those of arginine-vasopressin 1 and serotonin type 421 (Fig. 2).
Radiological evaluationThe radiological presentation is variable, from obvious cases with multiple macronodules in each adrenal gland, to cases that only have a 1–2cm macronodule in each adrenal gland. When only one nodule is seen in both adrenal glands without pathological diagnosis, the differential diagnosis between PBMAH and bilateral adrenal adenomas can be complex. Comprehensive image analysis is helpful in identifying additional features suggestive of PBMAH, such as the presence of smaller nodules visible in one adrenal gland or both adrenal glands (>2–3cm) and when there is no atrophy in the normal gland on the side of the nodule, which is suggestive of diffuse enlargement of the gland.2
Detailed radiological characterisation is a crucial part of the diagnosis in bilateral adrenal lesions with the primary goal of excluding malignancy.49 The differential diagnosis should be made mainly with other bilateral adrenal lesions. It is important above all to rule out adrenal metastases, infiltrative diseases, bilateral adrenal carcinoma and bilateral pheochromocytoma, although these last two conditions usually present more commonly with unilateral forms. The imaging modality of choice for radiological evaluation, and the most widely used, is computed tomography (CT) without contrast, which can provide information on the shape, size and density of the lesion. PBMAH nodules generally appear similar to adrenal adenomas: lipid-rich with a low attenuation value on non-contrast CT of less than 10 Hounsfield Units (HU), and with rapid contrast washout after iodinated contrast injection.58
TreatmentThere are three options for treatment: surgery, medical treatment or observation. The choice of treatment will depend mainly on the degree of hypercortisolism, the age of the patient and the presence or absence of comorbidities (Table 2).
Treatment options in patients with PBMAH.
| Therapeutic option | Indications | Hypercortisolism control |
|---|---|---|
| Cushing's syndrome | ||
| Bilateral adrenalectomy | Severe Cushing's syndrome | 100%59 |
| Unilateral adrenalectomy | Mild Cushing's syndrome, asymmetric adrenal involvement, suspicion of low adherence to GC treatment or high surgical risk | 55–95%61,62 |
| Blocking medical treatment | Aberrant receptor study positive for GIP, catecholamines, or LH-HCG | Variable, usually transient response8,9 |
| Steroidogenesis inhibitors | Contraindication to surgery or as pre-surgical treatment | 75.9% with metyrapone, 71% with ketoconazole,66 1 of 1 with osilodrostat67 |
| Autonomous cortisol secretion | ||
| Unilateral adrenalectomy | ACS with associated comorbidities, especially if they are poorly controlled | Improvement of comorbidities related to ACS, especially hypertension and diabetes10 |
| Blocking medical treatment | Aberrant receptor study positive for GIP, catecholamines, or LH-HCG | Variable, usually transient response8,9 |
| Medical treatment:- Metyrapone 250mg (6pm) and 500mg (10pm)- Ketoconazole 200–400mg/day- Mifepristone 200–400mg/day | ACS with associated comorbidities, especially if they are poorly controlled, and a patient with high surgical risk or who rejects unilateral adrenalectomy. | - Correction of circadian rhythm disturbance in 6/669- 1 case with normalisation of hypercortisolism70- Improved insulin resistance, AHT, quality of life and cardiometabolic parameters71–73 |
ACS: autonomous cortisol secretion; AHT: arterial hypertension; GIP: gastrointestinal inhibitory peptide.
In cases of overt Cushing's syndrome, the classic treatment is bilateral adrenalectomy, but it has the problem that the patient will need life-long treatment with gluco- and mineralocorticoids. On the other hand, it has the major advantage that there is no risk of recurrence if the resection is complete,59 and that cortisol excess is always sufficiently controlled.
Unilateral adrenalectomy would be reserved mainly for cases of ACS with associated ACS-related comorbidities10 or patients with overt Cushing's who are expected to have poor adherence to glucocorticoid replacement therapy or are at high surgical risk.60 Control rates of hypercortisolism are variable, up to 97% according to some series, with recurrence rates of 15–20%.61 However, more recent series reported recurrence rates of 55%, especially in patients with overt Cushing's syndrome, recommending that unilateral adrenalectomy should be reserved only for patients with mild Cushing's syndrome or ACS.62 After surgery, clinical improvement and control of comorbidities related to hypercortisolism, such as obesity, hypertension or diabetes are observed. Regarding the hormonal alterations associated with hypercortisolism, UFC levels normalise in most patients while the DST or the evolution of the circadian rhythm remain altered in a high percentage of cases.2 The lack of optimal control of hypercortisolism after the unilateral approach could explain why a recent study found that patients treated with unilateral adrenalectomy had higher mortality rates than those treated with bilateral adrenalectomy.63 The selection of the adrenal gland to be resected should be based on size, recommending resection of the largest adrenal gland.28 In addition, NP-59 scintigraphy can be helpful since in some cases the radiotracer uptake may be very asymmetric. Similarly, although adrenal venous sampling is useful to determining which adrenal should be removed in primary aldosteronism, its role in PBMAH is controversial. A recent study with 16 patients did not find evidence for recommending adrenal venous sampling for this purpose.
A level of UFC twice above the upper limit of normal and the presence of marked asymmetry of adrenal enlargement are the best predictors of cure of hypercortisolism after unilateral adrenalectomy, according to some authors.19 However, on the other hand, other studies indicate that patients with a contralateral adrenal volume of >33.54ml or a preoperative UFC level of >216.08mg/24h are more likely to have recurrence of hypercortisolism.62
New techniques have been proposed that try to avoid the risks of bilateral adrenalectomy and minimise the risk of recurrence that unilateral resection entails.64 In a prospective study, a total adrenalectomy of the largest adrenal gland and a contralateral partial adrenalectomy were performed. With this technique, 95% of patients achieve control of hypercortisolism and only one recurrence of hypercortisolism has been reported at 30 months of follow-up. Improvement in cardiometabolic parameters after surgery has also been observed.
Medical treatmentAn elegant way of treating cortisol excess in PBMAH is the option of blocking aberrant receptors in patients with a positive aberrant receptor study.65 The problem is that the response to these treatments is usually transient, although cases of long-term response have been described with the use of propranolol and GnRH agonists.8,9 However, the biochemical response is not always accompanied by a clinical response and sometimes intolerance to treatment occurs due to the need to use high doses of beta-blockers.8 Monitoring is easier in cases with Cushing's syndrome, in which the normalisation of UFC is a parameter of hypercortisolism control, while in cases of ACS it would be necessary to consider repeating the altered aberrant receptor test under blocking treatment.
Steroidogenesis inhibitors such as ketoconazole, metyrapone or osilodrostat could be used in those patients with Cushing's syndrome without aberrant receptors for whom surgery is contraindicated, or as pre-surgical treatment with the aim of achieving control of hypercortisolism before the intervention.53,66,67 While there is little documented experience, treatment with steroidogenesis inhibitors of patients with ACS could be a viable option. Chronotherapy, administration in a diurnal rhythm to restore the cortisol rhythm, could be a more physiologic approach and normalise not only UFC, but also late-night salivary cortisol. We have the same medical treatment options, but also the option of follow-up without specific treatment for hypercortisolism.68–73 The greatest experience exists with metyrapone. In a recent proof-of-concept study led by Debono, metyrapone was administered to six patients with ACS, with the first dose of 500mg administered at 6 p.m. and a second dose of 250mg at 10 p.m. This pattern led to a correction of the disturbance in the circadian rhythm of cortisol secretion in all six patients.69 With ketoconazole, there is less experience in ACS. A case report described long-term control of hypercortisolism for 10 years with low doses (200–400mg/day) of ketoconazole, leading to a significant improvement in the control of arterial hypertension.70 Improvements in glycaemic control and weight loss have also been reported with the use of the non-selective glucocorticoid antagonist mifepristone. Where mifepristone seems to be more useful is in patients with ACS and glycaemic disorders.71–73 Another selective glucocorticoid receptor antagonist, relacorilant,74 is under development for patients with cortisol-secreting adenomas or hyperplasia who have diabetes mellitus/impaired glucose tolerance and/or uncontrolled hypertension. Patient recruitment ended in August 2022 and results are expected soon. Another phase I clinical trial investigated the usefulness of 11β-hydroxysteroid dehydrogenase type 1 inhibitors in patients with adrenal Cushing's syndrome or ACS. Only 16 patients were included, but the study showed a significant improvement in glucose levels, a reduction in body mass index (BMI) and fat percentage, and an increase in muscle mass percentage.71
ConclusionsPBMAH accounts for less than 2% of cases of Cushing's syndrome. More commonly, PBMAH is found among patients with adrenal incidentalomas, often presenting with ACS. Up to 80% of cases demonstrate aberrant receptor expression, if appropriately investigated, which may be a potential target for pharmacological treatment, although its presence is not specific to PBMAH. Germline mutations in ARMC5 are the most common cause of hereditary PBMAH. Affected subjects have more severe clinical phenotypes, with a higher prevalence of overt Cushing's syndrome and larger adrenal glands than non-carriers of the mutation. The most recently described germline mutation is KDM1A, exclusively found in patients with aberrant receptors for GIP, and this mutation has also been associated with an increased risk of haematopoietic neoplasms. The treatment of choice in PBMAH is adrenalectomy, when overt Cushing's syndrome is present. Unilateral adrenalectomy should be considered instead of bilateral adrenalectomy, depending on the degree of hypercortisolism and morphologic asymmetry. Unilateral adrenalectomy is indicated in ACS with a prominent metabolic phenotype and/or hypercortisolism-associated comorbidities. In the rest of ACS cases, medical treatment or observation can be considered.
Ethical approvalAll procedures performed in the participants of the study were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The study was approved by the local ethics committee of the Ramón y Cajal Hospital (Approval date: 23 September 2019).
Financial supportNot applicable.
Conflict of interestThe authors have no conflict of interest.
