





The transition period from child to adult represents a crucial phase in the growth process where multiple physical and psychosocial changes occur. It has been arbitrarily defined as the period extending from late puberty to full adult maturity (i.e., from mid to late teenage years until 6–7years after achievement of final height).
The aim of this guideline is to emphasize the importance of adequate hormone replacement during this period and to review reassessment of pituitary function. In patients with GH deficiency diagnosed in childhood, an attempt is made to answer when to retest GH secretion, when to treat and how they should be monitored. Thyroxine, glucocorticoid, and sex steroid replacement is also reviewed.
Se entiende por periodo de transición del niño al adulto a una etapa de cambios físicos y psicológicos que, de forma arbitraria, se extiende desde el final de la pubertad hasta que la maduración adulta se completa. Comprende, habitualmente, los 6 a 7años posteriores al momento en que el niño adquiere la talla adulta.
Con esta documento pretendemos poner de manifiesto la importancia de la adecuada sustitución de los diferentes déficits hipotálamo-hipofisarios durante este período. Para ello revisamos la reevaluación del status hipofisario en los pacientes deficitarios durante la infancia. Tratamos de dar respuesta a las preguntas que pueden surgir y ofrecemos unas recomendaciones claras de cómo abordar la deficiencia de GH en este período. Posteriormente abordamos también la evaluación y la sustitución del eje adrenal, tiroideo y gonadal.
The transition period from child to adult is defined as a stage of physical and psychological changes arbitrarily considered to extend from late puberty to complete adult maturity. It usually comprises the six or seven years subsequent to the attaining of adult height by the child.1
The transition of patients with growth hormone deficiency from the pediatric to the adult endocrinology teamsAlthough the use of growth hormone (GH) during the transition period continues to be controversial, there is increasing interest in determining: (a) tissue maturation over time in GH-deficient and healthy adolescents; (b) the potential consequences of treatment discontinuation or “holidays”; and (c) the effect, if any, of GH replacement on fracture morbidity and cardiovascular disease. Only long-term follow-up in prospective studies can clarify these issues.
Growth vs body maturity. Current problems in the transition periodLongitudinal growth is considered to be completed when growth velocity is less than 1.5–2.5cm/year and/or bone maturation is 97–98%. These goals are usually achieved at a bone age of 14–15years in girls and 16–17 years in boys. In this situation, only a small residual capacity of longitudinal growth is left. However, body maturity—lean mass, fat, and bone mineral density (BMD)—is not complete yet, and may not occur in some cases until up to almost 30years of age. It is generally accepted that:
- •
Peak bone mass is achieved between 20 and 25years.
- •
Muscle mass increases even beyond 20years of age in males and 14years in females.
- •
Fat mass increases even beyond 20years of age in females and until the end of puberty in males.
- •
That is, females gain fat mass and males gain muscle mass after the end of puberty.
A comparison of equivalent groups of patients with adult-onset (AO), previously untreated GH deficiency and patients with child-onset (CO) GH deficiency adequately treated to the end of longitudinal growth shows that the latter have: a shorter height (−1 standard deviation [SD]), lower body mass index (BMI), 80% of the lean mass, fat, and BMD values in the AO group, and levels of insulin-like growth factor type1 (IGF-1) and insulin-like growth factor binding protein 3 (IGFBP3) 3 or 4SD lower as compared to the AO group.
That is, for a similar GH deficiency, significant differences are seen between the CO and AO groups, which may be due to a restriction of adult body maturity due to untreated GH deficiency during the transition period. Thus, the discontinuation of treatment with GH in boys with GH deficiency at the end of longitudinal growth is associated with: (a) decreased muscle strength and mass; (b) increased body fat, mainly in the abdomen; (c) the arrest or reversal of muscle mass and BMD gain, with decreased bone formation markers; and (d) lipid profile impairment and, predictably, the occurrence of the typical features of adult-onset GH deficiency, which may lead to increased cardiovascular risk.2–4
Body development is not complete at the end of growth, and the available studies show evidence suggesting that GH action is required in the post-pubertal transition phase to achieve normal adult status. CO patients are not and should not be considered as adults at the time of the final dose of GH at the end of growth. We recommend that they receive specific care and continue to receive treatment to complete body development.
The transition phase is not usually clearly included in published consensuses and guidelines on GH treatment in children and adults, and we therefore face:
- a)
Clinical problems: the literature on transition is characterized by a lack of information about:
- -
What the objectives of treatment with GH in this stage should be, and how previously deficient subjects should be re-evaluated (different clinical criteria).
- -
GH dosage.
- -
The actual benefits of GH replacement in late adolescence or early adulthood.
- -
- b)
Practical problems: the lack of communication between pediatric and adult endocrinology departments:
- -
This usually occurs with the transfer of care at adolescence.
- -
This has prevented a comparison of patients who continue treatment with GH (are transferred to adult endocrinology) with those who show a good response to GH on re-evaluation and do not undergo monitoring.5–7
- -
This has resulted in a scarcity of large studies using homogeneous criteria that might have made them comparable, and has caused some authors to question the benefits attributed to GH therapy in adulthood.
Objectives in the transition periodThe 2005 review of the European Society for Pediatric Endocrinology-EJE suggested the following objectives for the transition phase8:
- 1.
The re-evaluation of the etiology and status of all other pituitary axes.
- 2.
A GH treatment regimen.
- 3.
The achievement of complete adult body development.
- 4.
Sexual and reproductive maturity.
- 5.
The achievement of complete adult psychosocial development.
- 6.
Patient education on his/her condition.
The first question to be answered may possibly be: “Does GH deficiency persist?”6,9–11
Who should be re-evaluated?Re-evaluation is required in subjects with GH deficiency, except for pediatric indications for GH treatment in children with no GH deficiency (e.g. with Turner syndrome),12 because treatment after growth arrest is not indicated in these cases.
It should not be forgotten that 75% of isolated idiopathic GH deficiencies diagnosed in childhood have a normal response to stimulation tests when adult age is reached.
Ideally, all patients who have received treatment with GH should be followed up, even if they show no deficiency upon re-evaluation. However, these patients are not followed up at most centers because they are discharged. Studies defining what occurs with this subgroup of patients and so allowing them to be compared to deficient patients who continue treatment are lacking.7
When should re-evaluation be done?The adequate time for re-evaluation is at the end of longitudinal growth, as previously defined.
Who is responsible?The person responsible for re-evaluation should be defined, and assessment should ideally be made jointly by both the pediatric and adult endocrinologist. As this is not feasible in most cases, it appears logical that the physician in charge of the patient at the time that final height is reached (bone age, growth velocity) should normally be responsible for the re-evaluation.
How should re-evaluation be conducted?There is agreement in the literature that the treatment-free interval should not be less than one month (and up to three months1) and that all other hormone deficiencies should be corrected. The effect of oral estrogens on IGF-1 levels should be considered, even though their influence in the diagnosis of persistent GH deficiency has not been assessed.
GH levels return to baseline in about one week after treatment discontinuation, while a return to baseline levels of IGF-1, IGFBP3, and acid-labile subunit (ALS) may take 6–12months.6,10
Re-evaluation should include not only the somatotropic axis, and thus the need for continued GH treatment, but also all other axes, and the influence of GH discontinuation on the dosages of other treatments should be taken into consideration. Diagnosing hypogonadotropic hypogonadism may be difficult because of delayed bone age and the usual difficulties for differential diagnosis between delayed physiological puberty and hypogonadotropic hypogonadism.9
Three different procedural protocols have recently been reported:
A. In its 2005 consensus, the European Society for Pediatric Endocrinology (ESPE) did not consider it necessary to re-evaluate patients with congenital or acquired severe panhypopituitarism (three or more deficiencies).8 GH deficiency exists in 96% of adults with three hormone deficiencies and in 99% of those with four deficiencies, and the same occurs during transition. For all other cases, two risk groups for persistent GH deficiency were established, for which the recommendations for re-evaluation are different (Figs. 1 and 2).
- a)
High risk:
- •
Severe GH deficiency in childhood of a genetic cause (with or without other associated hormone deficiencies) or related to a structural hypothalamic–pituitary change, central nervous system tumors, or a history of high-dose cranial irradiation.
- •
In this situation, IGF-1 levels higher than −2SD are diagnostic of GH deficiency in adults. If IGF-1 levels are more than −2SD, a GH stimulation test should be performed.
- •
- b)
Low risk:
- •
Idiopathic GH deficiency, either alone or associated with other hormone deficiencies. In this case, IGF-1 measurement and a GH stimulation test are required. The possibility of evolving endocrine disease should however be assessed, and re-evaluation should be done in 6–12 months if the response to the insulin-induced hypoglycemia test for GH is higher than 5μg/L and less than 10μg/L.
- •

Proposal of the European Society for Pediatric Endocrinology for transition. GHD: GH deficiency.
Modified from Clayton et al.8

Proposal of the European Society for Pediatric Endocrinology for re-evaluation. GHD: GH deficiency. SD: standard deviation.
Modified from Clayton et al.8
Up to 75% of cases of isolated GH deficiency (IGHD) in childhood are not confirmed in re-evaluation, probably because they are partial deficiencies. However, the deficiency persists in adulthood in 25% of cases.7,10,13
B. Radovick and DiVall6 subsequently established a protocol consisting of three groups:
- a)
High risk:
This group includes patients with organic disease with multiple pituitary deficiencies (MPHD) or IGHD with mutation in genes influencing pituitary gland development (POU1F1, PROP-1, HESX-1, LHX-3, LHX-4) or GH gene expression (GH-1 mutation) or midline changes with MPHD. Re-evaluation is not required in these patients, and treatment may be continued with dosage adjustment.
- b)
Intermediate risk:
This group includes patients with idiopathic or acquired MPHD and acquired IGHD of unknown etiology, or with a history of tumor, surgery or pituitary irradiation. These patients should be re-evaluated after one month without treatment and, if their IGF-1 level is normal, a stimulation test (insulin-induced hypoglycemia or GHRH-arginine test) should be performed, as in the low risk group.
- c)
Low risk:
This group includes patients with IGHD with normal pituitary gland and no history of interest. They should be re-evaluated after at least one month without treatment, and a stimulation test should be performed in all of them.
This is actually a modification of the ESPE protocol.
C. Protocol of the American Association of Clinical Endocrinologists (AACE) 2009.14,15
This is the first protocol with levels of evidence and the most comprehensive one. It differs from prior protocols in that specifications are given about how to act in the event of an idiopathic lesion or a suspected hypothalamic origin, as well as in countries where GHRH is not available, so making a GHRH+arginine test difficult. The glucagon test is proposed as a third option, or as a second option after the insulin-induced hypoglycemia test when GHRH is not available. The importance of considering patient BMI in assessing response in the GHRH+arginine test is also stressed.
The ESPE guidelines are probably the ones most widely used in our environment. However, because of the above discussed aspects, the AACE guidelines may be considered as the most complete, and are the only ones which include levels of evidence.
IGF-1 measurements and stimulation testIGF-1After the discontinuation of GH treatment, it may take from 6 to 12months for IGF-1 levels to return to baseline.10 On the other hand, normal IGF-1 levels do not rule out GH deficiency in adults.16
The IGF-1 cut-off value is less than 84μg/dL for some authors,17 and less than −2SD (approximately 100μg/dL) for other authors.2
Causes of falsely low IGF-1 levels include malnutrition, liver disease, poorly controlled diabetes, and hypothyroidism.
It should also be noted that IGFBP-3 levels have no diagnostic value.
GH stimulation testTests to be used are not the same as those indicated for baseline assessment at pediatric age, because clonidine, l-dopa, or arginine tests alone are not helpful.
GHRH+arginine or GHRH+GHRP6 tests require the integrity of the hypothalamic–pituitary axis. Hence, in the first five years after hypothalamic radiotherapy, an insulin-induced hypoglycemia test should be considered if all other stimuli are normal.15,17
As regards the GH cut-off value, it should be noted that GH secretion is not the same at 16 and 50years, and it is therefore not logical to always use the same value. The ESPE proposes using for all stimuli a cut-off value of 5μg/L. A GH value less than 5.1μg/L with insulin is equivalent to a value less than 4.15μg/L with GHRH+arginine (95% sensitivity and 92% specificity for adult GH deficiency).5
Cut-off values therefore differ from those in AO. In this case, treatment for severe deficiency should be considered when GH levels are less than 3μg/L after the insulin-induced hypoglycemia and glucagon tests.18 Hypoglycemia continues to the gold standard, but shows high individual variability. In addition, it does not assess differences based on the BMI, and obese patients have a lower GH response. The lack of a BMI cut-off point has clinical implications. Moreover, in the reported studies,19,20 the control group also included obese subjects, but did not differentiate responses according to the BMI. On the other hand, normal or even elevated IGF-1 levels may be detected in simple obesity despite the presence of GH deficiency, because sensitivity may increase (greater IGF-1 response to low GH doses).
If ESPE criteria are used for the response to GH stimulation tests, there are three diagnostic categories8:
- -
GH deficiency in childhood: peak GH less than 10μg/L.
- -
GH deficiency in transition: peak GH less than 5μg/L.
- -
GH deficiency in adulthood: peak GH less than 3μg/L.
The cut-off point of GH less than 5μg/L is not fully accepted by the healthcare authorities. This is changing in some advisory committees on GH treatment, in which the protocols are being reviewed and this cut-off point for transition is already under consideration.13 On the other hand, some authors advocate a GH level of less than 6.1μg/L.16 In our view, the difference could lie in the laboratory method used for GH measurement. It is therefore important to be aware of the method used, because the cut-off points used in most publications are defined for radioimmunoassay (RIA).
The GHRH+arginine test is validated.9,21 It has the advantage of a low individual variability. It is at least as sensitive and helpful as the insulin-induced hypoglycemia test to re-evaluate GH deficiency in transition. It is only contraindicated in renal failure, and has a very good safety profile. There are no sex and age differences, but differences based on BMI are taken into consideration. Thus, GH levels suggesting deficiency are as follows: for BMI<25kg/m2, GH<11μg/L; for BMI 25–30, GH<8μg/L; and for BMI>40, GH<4μg/L.
When the GHRH+GHRP6 test is used, GH deficiency is defined as a value less than 10μg/L, but large studies using homogeneous criteria are lacking. If a value higher than 10μg/L and lower than 20μg/L is found, a repeat test should be performed.
The use of GHRH may cause side effects, including flushing, vasodilation, paresthesia. nausea, or altered taste. In addition, this test is not available in many centers.
Should all deficient patients be treated?The biochemical parameters have been discussed in the foregoing, but it should not be forgotten that final diagnosis should be established taking into consideration peak GH, IGF-1, symptoms, genetics (if applicable), and magnetic resonance imaging (MRI).
The 2005 ESPE consensus proposed that treatment should be offered to all deficient patients, while other more critical authors think that the decision should not only be based on a biochemical cut-off point, but that patients should be assessed integrally and their preferences should be taken into account after the potential advantages of treatment in adult age have been discussed with them. If the patient declines treatment, long-term follow-up should be performed.5
In cases of IGHD in childhood confirmed at re-evaluation, treatment is not approved by some advisory committees. We suggest that treatment be requested from the relevant committees, and if not approved, that the possibility of resubmission for assessment of treatment as “use outside indication” should be considered.
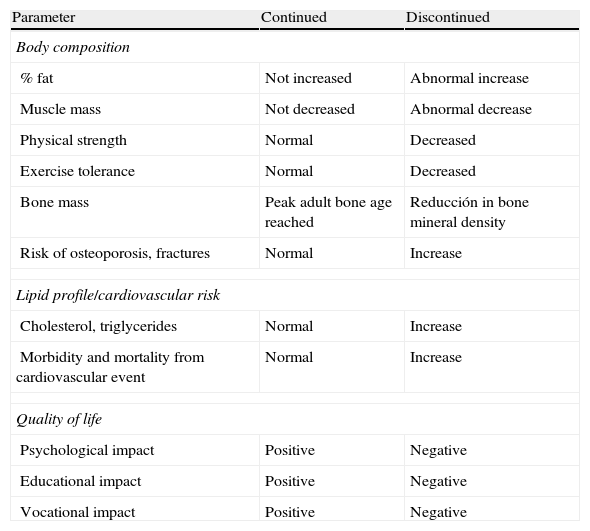
Table 1 shows differences in body composition. bone mass, lipid profile, and quality of life between patients who discontinue and continue treatment with GH after reaching adult height.1Table 2 shows the effects of GH replacement on body composition and bone.2–5,15
Risks versus benefits of GH therapy during transition.
| Parameter | Continued | Discontinued |
| Body composition | ||
| % fat | Not increased | Abnormal increase |
| Muscle mass | Not decreased | Abnormal decrease |
| Physical strength | Normal | Decreased |
| Exercise tolerance | Normal | Decreased |
| Bone mass | Peak adult bone age reached | Reducción in bone mineral density |
| Risk of osteoporosis, fractures | Normal | Increase |
| Lipid profile/cardiovascular risk | ||
| Cholesterol, triglycerides | Normal | Increase |
| Morbidity and mortality from cardiovascular event | Normal | Increase |
| Quality of life | ||
| Psychological impact | Positive | Negative |
| Educational impact | Positive | Negative |
| Vocational impact | Positive | Negative |
From Argente et al.1
Effects of GH replacement in the transition period.
| Body compositionIncrease in muscle massDecrease in fat massIncrease in bone massFemales gain a small amount of muscle mass with no change in fat massMales gain quite a lot of muscle mass and lose a significant amount of fat massChange in bone mass/height ratio is greater in males |
| In boneIn GH deficiency at the start of childhood there is a significant decrease in cortical thickness and bone mineral densityGH discontinuation in transition limits the acquisition of peak bone mass and promotes the development of osteopenia in adult ageTreatment with GH increases bone mineral content in cases with more severe GH deficiency |
| MetabolicGH deficiency in transition causes increases in total cholesterol, LDL cholesterol, and apoprotein B with HDL decrease. With replacement, changes vary depending on the authors: no change in LDL, HDL cholesterol, and TG, or increase in HDL or decrease in LDLTreatment with GH decreases insulin sensitivity (euglycemic clamp) despite simultaneous improvement in body composition |
| Quality of lifeThe only data with some predictive capacity is baseline quality of life. Sex, age, deficiency severity, other associated hormone deficiencies, body composition changes, or IGF-1 levels have no predictive value |
The convenience or not of a “holiday period” is another controversial issue, because no data are available on the effect of these short interruption periods on the metabolic state of patients during transition.
It should also be borne in mind that in other countries where treatment is not reimbursed, the lack of easily objectified immediate results (as we are no longer measuring things such as height) may make treatment continuity difficult.
Clinical documentationApproval by an advisory committee is traditionally required for treatment with GH and related substances. Most such committees are regional, after the devolution of healthcare responsibilities to the different Spanish autonomous communities. The protocols to be completed should include the transition period, taking into consideration the cut-off points for this period.
GH treatment regimenThe initial dosage was first calculated based on weight, then by ideal weight, and finally by approximation to the adult dose.
Once the final weight is reached, the pediatric dose of 25μg/kg/day should not be used, but an approximation to the adult dose should be used, maintaining normal IGF-1 (between 0 and +2SD), starting with 0.2–0.5mg/day. During the transition period, a dose of more than 2mg daily is rarely required.
ESPE recommendations advise starting with 0.2g/day in males and 0.3g/day in females.8 Healthy women secrete three times more GH than men, although they usually have similar serum IGF-I levels. In GH deficiency in adults, the exogenous GH dose required to achieve a given serum IGF-I level is significantly higher in females as compared to males (because of relative GH resistance, partly mediated by estrogens).
Treatment monitoringPossible interference with other hormone treatments should be borne in mind, and it should not be forgotten that a change in GH dosage may require the adjustment of the dosages of all other replacement treatments. Once puberty is completed, oral estrogen replacement should not be administered, because estrogens decrease GH action when given by this route, so that higher GH doses are required. Moreover, GH increases the conversion of T4 into T3, so that levothyroxine adjustment may be needed at treatment start. The hydrocortisone dose may also have to be increased due to GH action on 11-beta-hydroxysteroid dehydrogenase type1. It should also be noted that de novo treatment may unmask secondary hypothyroidism or secondary adrenal insufficiency.
Echocardiography is included in some protocols based on the potential improvement in cardiac contractility after treatment start shown in some studies. However, the guidelines state that echocardiography should only be performed if clinically indicated.14,15
Safety of treatmentThere is evidence that treatment with GH is safe and induces neither tumor growth nor the occurrence of new neoplasms, although it is contraindicated in patients with active neoplasms.15 No evidence exists of a greater growth rate of remnants or recurrences in patients with pituitary tumors or craniopharyngiomas. The risk of type1 or type2 diabetes mellitus is not increased either.15
Conclusions and recommendationsThe adequate transition of adolescents with GH deficiency from pediatric to adult endocrinology departments represents a challenge.
In any case, each patient should be considered individually even when published guidelines are being followed.
The following recommendations should be taken into consideration:
- 1.
We recommend treatment with GH during the transition period, because for a similar GH deficiency, significant differences have been noted between CO and AO, which may be due to periods without treatment at a crucial time for the acquisition of an adequate body composition.
- 2.
We recommend close collaboration between pediatric and adult endocrinology departments in the joint assessment of these patients.
- 3.
All cases of GH deficiency should be re-evaluated, except for pediatric indications of GH treatment in children with no GH deficiency.
- 4.
Re-evaluation should be performed at the end of the longitudinal growth period.
- 5.
The treatment-free interval before re-evaluation should be at least one month, and all other hormone deficiencies should be corrected.
- 6.
Patients with organic pathology or having midline changes with MPHD, or patients with IGHD having mutations in genes influencing pituitary gland development or GH gene expression require no re-evaluation, and treatment can be continued with adjustment of the dosage.
- 7.
Patients with idiopathic or acquired MPHD and acquired IGHD of unknown etiology, or with a history of tumor, surgery or pituitary irradiation. should be re-evaluated after one month without treatment, and if the IGF-1 level is normal, a stimulation test should be performed.
- 8.
Patients with IGHD and a normal pituitary gland and no history of interest should be re-evaluated after at least one month without treatment, and a stimulation test should be performed in all of them.
- 9.
The insulin-induced hypoglycemia test is currently recommended because it continues to be the reference method. When this test is contraindicated, the glucagon test may be used instead.
- 10.
The GHRH+arginine or GHRH+GHRP6 tests require the integrity of the hypothalamic–pituitary axis. The GHRH+arginine test has the advantages of a low individual variability and a different cut-off point depending on the BMI.
- 11.
During the transition period, a GH cut-off point less than 5.1μg/L with insulin should be considered, which is equivalent to a lower value of 4.15μg/L with GHRH+arginine.
- 12.
The measurement of IGF-1 may be helpful. It should be taken into account that it may take some time for IGF-1 levels to return to baseline after the discontinuation of GH treatment. The cut-off IGF-1 value is established at less than −2SD (approximately 100μg/dL).2 The possible causes of falsely low levels should be kept in mind.
- 13.
The starting dose should be 0.2–0.5g/day. Possible interference with other hormone treatments should be taken into consideration, and it should not be forgotten that any change in GH dosage may require the adjustment of the dosages of all other replacement treatments.
- 14.
IGF-1 should be measured at six weeks of treatment start to adjust treatment in order to maintain IGF-1 in the 25th to 75th percentile of the normal range. After adjustment, IGF-1 should be measured every three months. To assess any change in dosage, IGF-1 should be measured at least six weeks later. Weight, BMI, waist/hip ratio. blood pressure, pulse, and quality of life should be assessed annually. Lipid profile tests and densitometry, with the T-score being assessed in the third decade of life, and the T and Z-scores subsequently, should be performed every two to five years.
As in any other stage, evaluation should be performed whenever the clinical data suggest that hypopituitarism might exist. Evaluation is also required in cases where, despite an absence of characteristic symptoms, there is a history of conditions that may cause the involvement of the hypothalamic–pituitary axis, such as tumors, inflammatory or infectious conditions, vascular disorders, trauma, irradiation, autoimmune and genetic changes, or others.
Re-evaluation of all other hypothalamic–pituitary axes during the transition period in patients who had or had not started treatment in childhood has not been studied so extensively as in GH deficiency. It should be stressed, however, that in patients with hypopituitarism, re-evaluation of all other axes should be performed, and the time of referral to endocrinologists experienced in the management of hypothalamic–pituitary disease in young adults should be determined.9
ACTH deficiency. Secondary or central adrenal insufficiencyIn children with hypopituitarism, assessment of the hypothalamic–pituitary–adrenal axis is crucial for long-term survival. The results of the initial evaluation may sometimes be normal, followed by function decrease or loss some years later.10 Approximately one third of patients with a normal response in the initial evaluation have developed adrenal insufficiency by the end of puberty.22
To assess ACTH secretion, plasma cortisol should be measured between eight and nine in the morning. The results should be assessed using the standard criteria for adrenal axis assessment.
The insulin-induced hypoglycemia test concomitantly used to assess GH and the CRH (corticoctoprin releasing hormone) test are the best tests for assessing the presence of central adrenal insufficiency. The glucagon test may also be helpful in children. The metyrapone test, as valid as the insulin-induced hypoglycemia test, has fallen out of use due to its unavailability.9 The 1μg ACTH test (low dose) was superior to the standard 250μg test for the diagnosis of secondary adrenal insufficiency in the Kazlauskaite et al. meta-analysis.23 It may have limitations, however. On the one hand, it has not been validated in patients with acute diseases, sleep-wake cycle disorders, or acute hypothalamic–pituitary changes, such as occur one month after surgery. The effect of food or fluid intake has not been validated either. Moreover, the value of the test may decrease when it is performed in the evening. On the other hand, no information is available in patients with low protein levels, because circulating cortisol binds with a high affinity to proteins. Preparation problems also exist, which may lead to false positive results (falsely low cortisol levels at 30min).23 To avoid such problems, a careful reconstitution method and additional steps should be used to prevent the adhesion of the preparation.24 It should also be noted that treatment with GH has variable effects on cortisol response to ACTH. The interruption period of GH treatment for re-evaluation could therefore be a good time for reviewing the function of the hypothalamic–pituitary–adrenal axis.25
TSH deficiency. Secondary or central hypothyroidismThis may be due to a deficient production of TSH or thyrotropin releasing hormone. It is diagnosed when free T4 levels are decreased with low or normal TSH levels.26 Basal TSH, while helpful for the diagnosis of primary hypothyroidism, does not allow us to rule out the presence of central hypothyroidism. TSH measurement after stimulation with TRH was traditionally used for diagnosis. This test also allowed for differentiating central hypothyroidism of a pituitary origin, with absence of response, from hypothyroidism of a hypothalamic origin, in which a delayed increase in TSH levels occurs. However, other diagnostic methods are now required because of the low current availability of TRH.9 One possibility in children would be to observe the nocturnal TSH peak (with a mean 114% increase under normal conditions), associated with free T4 levels in the lower third of the normal range or even lower, which may be more sensitive than the TRH test.9,26
It has been suggested that GH replacement therapy may cause central hypothyroidism in children with GH deficiency with discretely low T4 levels. However, studies where nocturnal TSH peak was measured found no thyroid function changes with GH treatment, unless central hypothyroidism already existed before treatment.26
During the transition period, the re-evaluation of free T4, T3, and TSH 6–12 weeks after treatment withdrawal could be useful in patients with no structural hypothalamic–pituitary disease or no deficiencies other than those of GH and TSH. If results suggest that thyroid function is normal and GH treatment is to be resumed, free T4 levels should be closely monitored, and treatment may be restarted if levels diagnostic of central hypothyroidism are found.9
Gonadotropin deficiency. Hypogonadotropic hypogonadismDelayed puberty is defined as the failure to start breast development and pubic hair appearance at 13years in girls and testis growth and pubic hair appearance at 13years in boys. It should be noted that this definition is only valid if chronological age agrees with bone age. However, in patients with GH deficiency bone age may not agree with chronological age and the age of definition may be delayed, and in some cases, the diagnosis is made in the transition period.9
Overall, spontaneous puberty may be assumed to be unlikely if: (a) no signs of puberty exist at a bone age of 13years in girls and 14 years in boys; (b) prepubertal levels of luteinizing and follicle-stimulating hormones, as measured by ultrasensitive methods, still exist; (c) prepubertal levels of estradiol in girls and testosterone in boys still exist; and (d) gonadal size is still prepubertal. To assess these data, ultrasound examination is required in girls. In such cases, the start of hormone replacement therapy is based on chronological age, emotional maturity, potential height, and bone density.9
Treatment of deficiencies in all other anterior pituitary hormones during transitionACTH deficiency. Secondary or central adrenal insufficiencyThis is the first deficiency requiring-urgent-replacement when suspected, because it may compromise the patient's life. The administration of an adequate steroid dose during the transition stage is crucial. Both the use of suboptimal doses that may cause hypoglycemia or even an adrenal crisis27 and underdosing, which may cause growth arrest, obesity, muscle weakness, and osteopenia, should be avoided.28
The most commonly used glucocorticoid is hydrocortisone (Hidroaltesona®, as 20mg tablets). This is a preparation with a short half-life which, when given as two or three daily doses, makes it possible to mimick the circadian rhythm of cortisol. Preparations with a longer half-life such as prednisone, prednisolone, or dexamethasone are less commonly used.
Doses equivalent to daily cortisol production, ranging from 12 to 14mg/m2/day, usually 15–20mg/day, should be used.29,30 During the transition stage, administration in three divided doses daily is recommended to avoid the peak that occurs after the morning dose and low levels during the night, as is usual with regimens of two daily doses.31,32 Plenadren® is a modified release hydrocortisone that mimics the physiological rhythm of cortisol secretion when given as a single daily dose. It is not marketed in Spain, but is available for use as a foreign drug. However, it is currently restricted to use in adrenal insufficiency in adults, and no data are available regarding its use in the transition phase.
In the event of mild to moderate stress due to a concomitant condition (fever, infection, minor surgery), the patient should be given double the standard dose (24mg/m2/day). If there is any condition preventing oral treatment, hydrocortisone must be administered by the intramuscular or intravenous route (Actocortina® 50–100mg by the parenteral route, to be repeated at 8–12h based on the clinical course).
In patients undergoing major stress, such as surgery or burns, hydrocortisone should be administered by the parenteral route, usually as 100mg of Actocortina® every 6–8h.
Treatment monitoring should be based on clinical and biochemical data, as there is no hormone marker that makes it possible to assess whether the administered dosage is adequate.
TSH deficiency. Secondary or central hypothyroidismTSH deficiency is treated with oral levothyroxine as a single daily dose, preferably in the morning and while fasting. It should be stressed that treatment with levothyroxine should never be started before ACTH deficiency has been ruled out, because it could trigger an adrenal crisis.
Treatment is essential for both the maturation and intellectual development of the patient but, as occurs with ACTH deficiency, overdosing should be avoided in order not to compromise growth and condition final height.
The total dose per kilogram of weight in the transition stage is approximately 2μg/day, lower than the requirements in childhood but higher than those in adult age. Treatment monitoring is based on the measurement of free T4. The dose is gradually increased every 8–10 weeks until free T4 level is maintained at the midpoint of the normal range.
Gonadotropin deficiency. Hypogonadotropic hypogonadismFemalesThe aim of treatment is to achieve normal pubertal development, and should be started at around 12years of age to induce puberty.
Treatment consists of estrogens. Two oral administration forms (ethinyl estradiol and estradiol valerate) and one transdermal administration form (estradiol patches) are available.
Treatment should be started with very low doses, ranging from an eighth and a tenth of the standard dose in adults, to promote bone mass gain and avoid compromising longitudinal growth.33 Ethynyl estradiol is the estrogen most widely recommended as the initial drug, and especially for the induction of secondary sexual characteristics.34 The starting dose is 2.5–5μg/day of ethinyl estradiol33 or estradiol patches of 25μg/day.
The dosage is subsequently doubled every 6 months for the following three years until the dose used in adult women is reached (20–35μg/day of ethinyl estradiol, 2–4mg of oral estradiol, or 50–100μg/day of transdermal estradiol35).
Once breast development is complete and/or after the first menses, progestogens should be cyclically associated in the last seven days of the cycle to achieve withdrawal bleeding and prevent endometrial cancer.
Transdermal patches are preferred to oral preparations for maintenance, because the latter have been related to impaired metabolic profile and body composition. Moreover, they block IGF-1 synthesis in the liver, a factor which should be taken into account in patients receiving GH replacement therapy. When the oral route is used, the minimum possible estrogen dose should be given.36,37
When induction of ovulation is desired, treatment consists of gonadotropins such as recombinant FSH (75–150IU daily) until the follicles reach a size of 16–18mm or the estrogen level is 200-pg/mL or higher, at which time hCG (5000IU) should be administered to induce ovulation in 36h. Ultrasound monitoring is essential. Treatment with pulsatile GnRH is only useful when the secretory capacity of FSH and LH is preserved, but it provides no advantages.
MalesReplacement is performed using testosterone derivatives. Parenteral and transdermal preparations are available.
Transdermal preparations include patches (containing 15–30mg) and gels (containing 25 and 50mg). They are both applied daily, and maintain normal and stable testosterone levels for 24h.
Preparations for parenteral administration include testosterone enantate and cypionate (as 100 and 250mg formulations), which should be administered every 14–28days, and testosterone undecanate (as a 1000mg formulation). The latter is a preparation with a long half-life providing stable testosterone levels over 12weeks.
In adolescence, parenteral testosterone enantate or cypionate are the drugs of choice. The starting dose is 25–50mg every four weeks (by the intramuscular route), and it is gradually increased every six to 12months up to 250mg every two to four weeks.33 This scheme has been shown to be effective for the induction of secondary sexual characteristics, and promotes linear growth without compromising bone maturation. Testosterone levels allow for treatment monitoring, and should be close to the midpoint of the normal range. If patches or gels are used, doses are 5 and 50g/day respectively.
HCG (500–1000IU 2–3times weekly) has traditionally been used to induce fertility, or to increase testicular volume in boys. If no adequate response occurs after 12weeks of treatment, recombinant FSH may be added (75–100IU three times weekly). There is however no evidence to show that such treatment restores or improves spermatogenesis. The administration of GnRH pulses has no advantages over the use of gonadotropins and is more expensive.
Conflicts of interestE. Fernández and J. M. Recio have no conflicts of interest. I. Bernabeu states that he has received fees as a lecturer from Pfizer and Ipsen. C. Fajardo and C. Álvarez Escolá state that they have received fees for participating as lecturers or moderators in meetings organized by Pfizer, Lilly, Novo Nordisk, and Ipsen.
We would like to thank the members of the Knowledge Area of Neuroendocrinology of SEEN who reviewed this manuscript, as well as Dr. Elías Delgado, vice-chairman of SEEN and coordinator of Knowledge Areas and Working Groups, for his suggested changes and for obtaining the support of the SEEN Board of Directors for study publication.
Please cite this article as: Álvarez-Escolá C, Fernández-Rodríguez E, Recio-Córdova JM, Bernabéu-Morón I, Fajardo-Montañana C, en representación del Área de Conocimiento de Neuroendocrinología de la Sociedad Espa¿nola de Endocrinología y Nutrición. Documento de consenso del área de conocimiento de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición para el abordaje del hipopituitarismo durante la transición. Endocrinol Nutr. 2014;61:68.e1–68.e11.
