



Se entiende por periodo de transición del niño al adulto a una etapa de cambios físicos y psicológicos que, de forma arbitraria, se extiende desde el final de la pubertad hasta que la maduración adulta se completa. Comprende, habitualmente, los 6 a 7años posteriores al momento en que el niño adquiere la talla adulta.
Con esta documento pretendemos poner de manifiesto la importancia de la adecuada sustitución de los diferentes déficits hipotálamo-hipofisarios durante este período. Para ello revisamos la reevaluación del status hipofisario en los pacientes deficitarios durante la infancia, tratamos de dar respuesta a las preguntas que pueden surgir y ofrecemos unas recomendaciones claras de cómo abordar la deficiencia de GH en este período. Posteriormente abordamos también la evaluación y la sustitución del eje adrenal, tiroideo y gonadal.
The transition period from child to adult represents a crucial phase in the growth process where multiple physical and psychosocial changes occur. It has been arbitrarily defined as the period extending from late puberty to full adult maturity (i.e., from mid to late teenage years until 6-7years after achievement of final height).
The aim of this guideline is to emphasize the importance of adequate hormone replacement during this period and to review reassessment of pituitary function. In patients with GH deficiency diagnosed in childhood, an attempt is made to answer when to retest GH secretion, when to treat and how they should be monitored. Thyroxine, glucocorticoid, and sex steroid replacement are also reviewed.
Se entiende por periodo de transición del niño al adulto a una etapa de cambios físicos y psicológicos que, de forma arbitraria, se extiende desde el final de la pubertad hasta que la maduración adulta se completa. Comprende, habitualmente, los 6 a 7 años posteriores al momento en el que el niño adquiere la talla adulta1.
Transición de pacientes con déficit de hormona de crecimiento entre los equipos de endocrinología pediátrica y endocrinología de adultosA pesar de que el uso de hormona de crecimiento (GH) durante el período de transición sigue siendo controvertido, existe un interés creciente en determinar: a)la evolución de la maduración tisular en los adolescentes deficitarios y los sanos; b)las posibles consecuencias de la interrupción del tratamiento o «período de vacaciones», y c)el efecto, si existiera, de la sustitución con GH en la morbilidad por fracturas y la enfermedad cardiovascular. Probablemente, solo el seguimiento a largo plazo en estudios prospectivos podría dar respuesta a estos interrogantes
Crecimiento vs maduración corporal. Problemas actuales en la transiciónEl crecimiento longitudinal se considera terminado cuando la velocidad de crecimiento es menor a 1,5-2,5cm/año y/o la maduración ósea es del 97-98%. Estos objetivos suelen alcanzarse con una edad ósea de los 14 a 15años en las niñas y de los 16 a 17años en los niños. En esta situación solo se conserva una pequeña capacidad de crecimiento longitudinal residual. Sin embargo, la maduración corporal —masa magra, grasa y densidad mineral ósea (DMO)— no es aún completa, pudiendo en algún caso demorarse hasta casi los 30años. En general se acepta que:
- •
El pico de masa ósea se alcanza entre los 20 y los 25años.
- •
La masa muscular aumenta incluso hasta más allá de los 20años en los varones y hasta los 14en las mujeres.
- •
La masa grasa aumenta incluso hasta más allá de los 20años en las mujeres y hasta el final de la pubertad en los hombres.
- •
Es decir, tras la finalización de la pubertad las mujeres ganan masa grasa y los hombres masa muscular.
Si comparamos grupos equivalentes de pacientes deficitarios de GH, por un lado los de inicio en la edad adulta (adult onset, AO) nunca tratados y, por otro, los de inicio en la infancia (child onset, CO) adecuadamente tratados hasta el final del crecimiento longitudinal, se observa que los CO tienen: una talla menor (–1 desviación estándar [DE]), un índice de masa corporal (IMC) más bajo, un 80% de la masa magra, grasa y DMO respecto a los de AO y concentraciones de factor de crecimiento similar a la insulina tipo1 (IGF-1) y de proteína3 de unión al factor de crecimiento parecido a la insulina (IGFBP3) de 3 o 4DE por debajo que los de AO.
Es decir, para un déficit similar de GH hay diferencias importantes entre CO y AO que pueden deberse a la limitación de la maduración corporal adulta por déficit de GH no tratado durante el periodo de transición. Así, la retirada del tratamiento con GH en niños deficitarios al final del crecimiento longitudinal se acompaña de: a)disminución de fuerza y de masa muscular; b)aumento de grasa corporal, fundamentalmente abdominal; c)detención o retroceso en la ganancia de masa muscular y de la DMO, con disminución de marcadores de formación ósea, y d)deterioro del perfil lipídico y previsiblemente de la aparición de las características típicas del déficit de GH del adulto que podrían llevar a un aumento del riesgo cardiovascular2–4.
El desarrollo somático no está completo al finalizar el crecimiento y los estudios disponibles muestran evidencias que indican que la acción de GH es necesaria en la fase de transición pospuberal para alcanzar el status normal adulto. Los pacientes con CO no son y no deben ser considerados adultos con la última dosis de GH al final del crecimiento. Recomendamos que reciban cuidados específicos y continúen recibiendo tratamiento para completar el desarrollo somático.
La fase de transición no suele estar claramente incluida en los consensos y guías publicadas sobre el tratamiento con GH en niños o en adultos, por lo que nos enfrentamos a:
- a)
Problemas clínicos: la literatura sobre la transición se ha caracterizado por la falta de información sobre:
- -
Cuáles deben ser los objetivos del tratamiento con GH en esta etapa y cómo reevaluar a los previamente deficitarios (distintos criterios clínicos).
- -
Dosis de GH.
- -
Beneficios reales de la sustitución con GH en la adolescencia tardía o inicio de la etapa adulta.
- -
- b)
Problemas prácticos: falta de comunicación entre servicios de pediatría y de endocrinología de adultos:
- -
Suele coincidir con la transferencia de cuidados al adolescente.
- -
Esto ha impedido comparar a los pacientes que continúan tratamiento con GH (pasan a endocrinología de adultos) con los que, al ser reevaluados, presentan buena respuesta a GH y no se realiza seguimiento5-7.
- -
Esto lleva consigo una escasez de estudios amplios y de criterios homogéneos que puedan hacerlos comparables y provoca que algunos autores cuestionen los efectos beneficiosos atribuidos al tratamiento con GH en la edad adulta.
Objetivos en el periodo de transiciónEn la revisión de Sociedad Europea de Endocrinología Pediátrica-EJE publicada en 2005 se plantearon como objetivos en la fase de transición8:
- 1.
Reevaluar la etiología y la situación del resto de ejes hipofisarios.
- 2.
Régimen de tratamiento con GH.
- 3.
Alcanzar el desarrollo somático adulto completo.
- 4.
Maduración sexual y reproductiva.
- 5.
Alcanzar el desarrollo psicosocial adulto completo.
- 6.
Educación del paciente respecto a su patología.
Quizá la primera pregunta que debemos contestar es: «¿Persiste el déficit de GH?»6,9–11.
¿A quién reevaluar?Se deben reevaluar los casos de déficit de GH, a excepción de las indicaciones pediátricas de tratamiento con GH en niños no deficitarios de GH (por ejemplo, con síndrome de Turner)12, ya que en estos casos el tratamiento posterior al cese de crecimiento no está indicado.
No debemos olvidar que el 75% de los déficits aislados de GH idiopáticos diagnosticados en la infancia tienen una respuesta normal a los test de estímulo al llegar a la edad adulta.
Lo ideal sería seguir a todos los que han recibido tratamiento con GH, aunque en la reevaluación no resultaran deficitarios. Sin embargo, el seguimiento en estos casos no se realiza en la mayoría de los centros ya que estos pacientes son dados de alta. En este sentido faltan publicaciones que definan qué ocurre con este subgrupo de pacientes y que permitan compararlos con los deficitarios que mantienen tratamiento posteriormente7.
¿Cuándo reevaluar?El momento indicado es al final del periodo de crecimiento longitudinal, según lo definido previamente.
¿Quién es el responsable?El responsable de la reevaluación debe definirse, y lo ideal sería que la valoración se realizara conjuntamente entre el endocrinólogo pediátrico y el de adultos. Como esto no es factible en la mayoría de los casos, parece lógico que una vez alcanzada la talla final sea el facultativo que esté atendiendo al paciente cuando este alcance su talla final (edad ósea, velocidad de crecimiento) quien inicie la reevaluación.
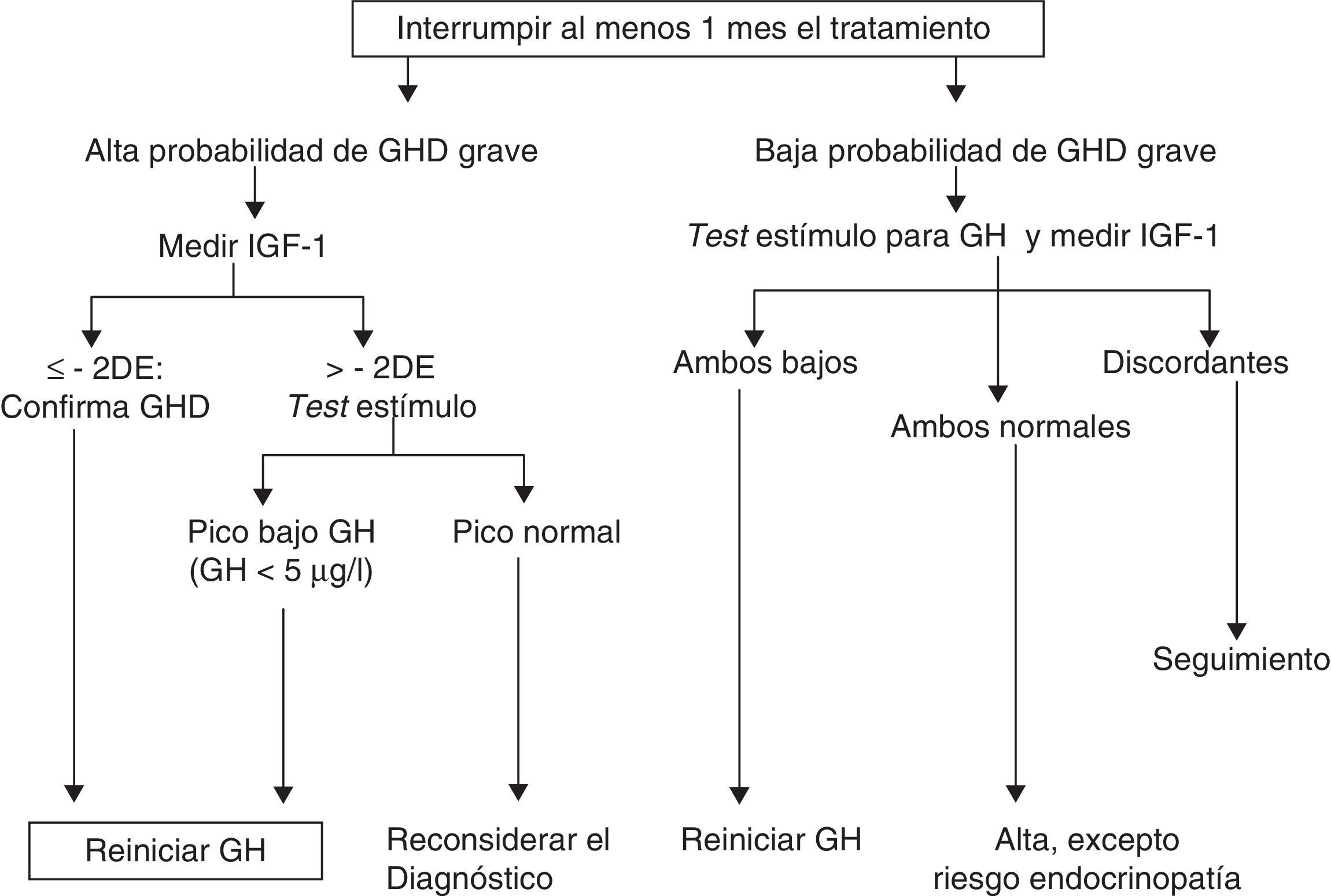
¿Cómo reevaluar?Existe acuerdo en la literatura en que el intervalo sin tratamiento no debe ser inferior a un mes (y hasta 3meses1) y que el resto de los déficits hormonales deben estar corregidos. Debe considerarse el efecto de los estrógenos por vía oral en los niveles de IGF-1, si bien su influencia en el diagnóstico de déficit de GH persistente no ha sido evaluada.
Los niveles de GH vuelven a su situación basal tras la suspensión del tratamiento en alrededor de una semana, pero sin embargo IGF-1, IGFBP3 y la subunidad ácido lábil (ALS) pueden tardar entre 6 y 12meses6,10.
Además del eje somatotropo, y por tanto la necesidad de continuar el tratamiento con GH, se deben reevaluar el resto de ejes y tener en cuenta la influencia de la suspensión de GH sobre la dosis de otros tratamientos. El diagnóstico de hipogonadismo hipogonadotropo puede ser difícil por el retraso de edad ósea y las dificultades habituales en el diagnóstico diferencial entre pubertad retrasada fisiológica e hipogonadismo hipogonadotropo9.
Podemos diferenciar 3 protocolos de procedimiento descritos recientemente;
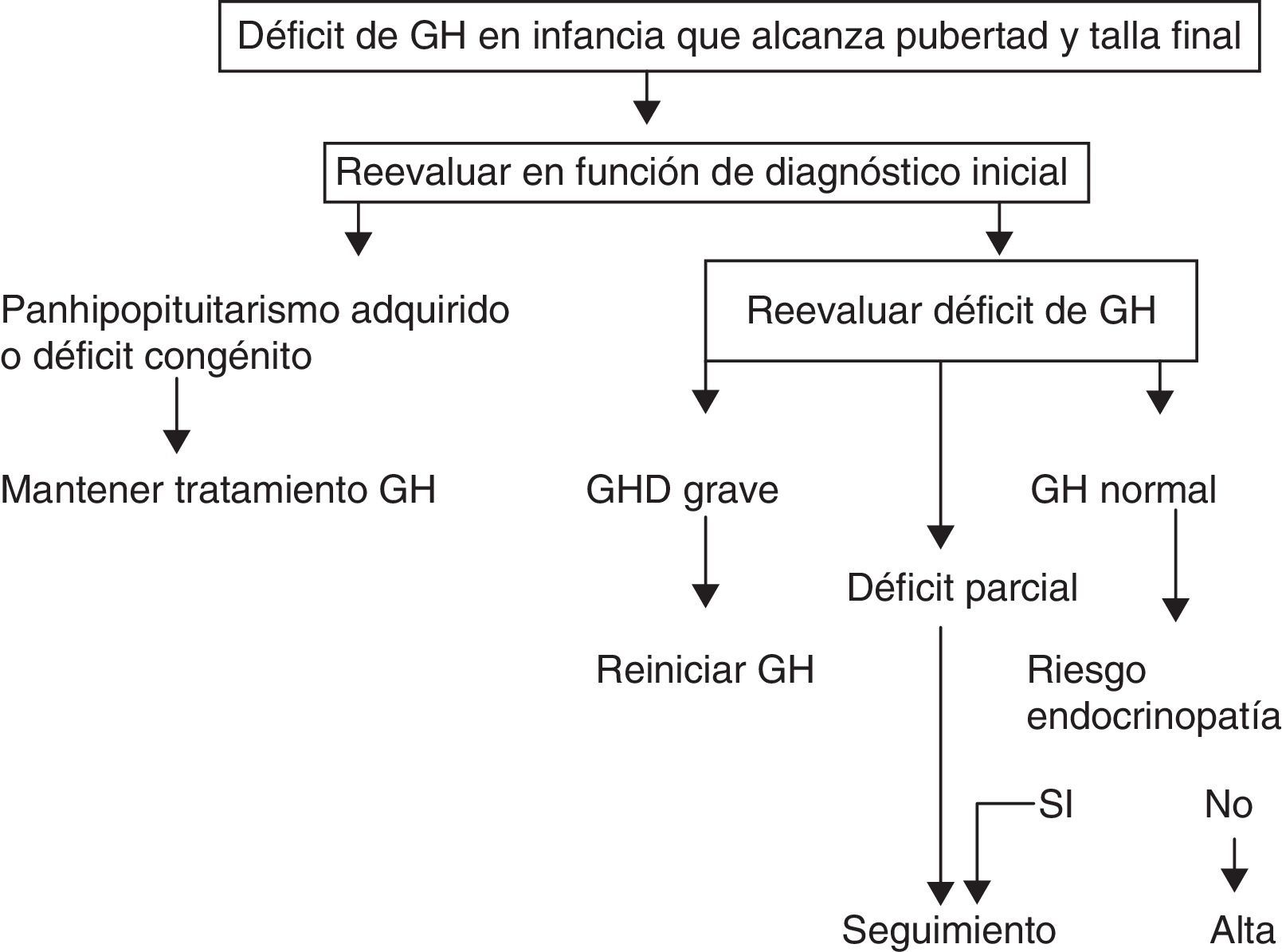
A. La Sociedad Europea de Endocrinología Pediátrica (ESPE) en el consenso de 2005 no consideró necesario reevaluar a los pacientes con panhipopituitarismo grave (3 o más déficits) congénito o adquirido8. En adultos con 3 déficits hormonales existe déficit de GH en el 96% de los casos, y en caso de 4 asciende hasta el 99%, y eso es similar en la transición. En los demás casos, estableció 2 grupos de riesgo de persistencia del déficit de GH en los que la recomendación de reevaluación es distinta (figs. 1 y 2).
- a)
Alto riesgo:
- •
Déficit de GH grave en la infancia de causa genética (con o sin otros déficits hormonales asociados) o bien en relación con una alteración estructural hipotálamo-hipofisaria, tumores del sistema nervioso central o con el antecedente de irradiación craneal a dosis altas.
- •
En esta situación una IGF-1 mayor de –2DE es diagnóstica de déficit de GH en el adulto. Si la IGF-1 es mayor de –2DE, debe realizarse un test de estímulo de GH.
- •
- b)
Bajo riesgo:
- •
Déficit de GH idiopático, ya sea aislado o asociado a otros déficits hormonales. En este caso se precisa IGF-1 y un test de estímulo para GH. Se debe valorar en todo caso la posibilidad de una endocrinopatía evolutiva y reevaluar en 6 a 12meses si la respuesta al test de hipoglucemia insulínica de GH fuera superior a 5μg/l e inferior a 10μg/l.
- •

Propuesta de la Sociedad Europea de Endocrinología Pediátrica para la transición. GHD: déficit de GH.
Modificado de Clayton et al.8.

Propuesta de la Sociedad Europea de Endocrinología Pediátrica para la reevaluación. GHD: déficit de GH; DE: desviación estándar.
Modificado Clayton et al.8.
Hasta el 75% de los casos de déficit aislado de GH (IGHD) de la infancia no se confirman en la reevaluación, probablemente por tratarse de déficits parciales. No obstante, en el 25% de los casos el déficit persiste en la edad adulta7,10,13.
B. Posteriormente Radovick y DiVall6 establecieron un protocolo en el que consideraron 3 grupos:
- a)
Riesgo alto:
En este grupo se incluyen pacientes con patología orgánica con múltiples déficits hipofisarios (MPHD) o IGHD con mutaciones en genes que influyen en el desarrollo de la hipófisis (POU1F1, PROP-1, HESX-1, LHX-3, LHX-4) o en la expresión del gen de la GH (mutación GH-1) o alteraciones de la línea media con MPHD. Estos pacientes no precisan reevaluación y el tratamiento podría mantenerse ajustando dosis.
- b)
Riesgo medio:
Se incluyen aquellos con MPHD idiopático o adquirido e IGHD adquirido de etiología desconocida o bien con antecedentes de tumor, cirugía o irradiación hipofisaria. Deben reevaluarse tras un mes sin tratamiento y, si la IGF-1 fuera normal, realizar un test de estímulo (hipoglucemia insulínica o test de GHRH-arginina), al igual que en los de riesgo bajo.
- c)
Riesgo bajo:
Los casos con IGHD con hipófisis normal y sin antecedentes de interés. Deben reevaluarse tras al menos un mes sin tratamiento y realizar en todos los casos test de estímulo.
Realmente es una modificación del protocolo de ESPE.
C. Protocolo de la American Association of Clinical Endocrinologists (AACE) 200914,15.
Es el primer protocolo con niveles de evidencia y el más completo. Como novedades respecto a los previos especifica cómo actuar en caso de lesión idiopática o sospecha de origen hipotalámico y ante la falta de suministro en algunos países de GHRH, lo que dificulta la realización de test de GHRH+arginina. Propone el test de glucagón como tercera opción de test o segunda opción tras el de hipoglucemia insulínica cuando no se dispone de GHRH. Además, se refuerza la importancia de considerar el IMC del paciente para valorar la respuesta en el test de GHRH+arginina.
Probablemente la guía de la ESPE sea la más empleada en nuestro medio. Pero por los aspectos anteriormente comentados, la guía de la AACE puede considerarse la más completa y la única en considerar niveles de evidencia.
Determinaciones de IGF-1 y test de estímuloIGF-1La IGF-1, tras la suspensión del tratamiento con GH, puede tardar en alcanzar su nivel «basal» entre 6 y 12meses10. Por otro lado, una IGF-1 normal no excluye el déficit de GH en adultos16.
Mientras unos autores establecen el punto de corte de IGF-1 inferior a 84μg/dl17, otros lo sitúan en IGF-1 menor a –2DE (aproximadamente 100μg/dl)2.
Se deben recordar las causas de IGF-1 falsamente bajas: malnutrición, enfermedad hepática, diabetes mal controlada e hipotiroidismo.
Hay que tener en cuenta también que los niveles de IGFBP-3 no tienen valor para el diagnóstico.
Test de estímulo de GHLos tests que debieran emplearse no son los mismos que los indicados en la valoración inicial en la edad pediátrica., ya que los de clonidina, L-dopa o arginina aislada no resultan útiles.
Los test de GHRH+arginina o GHRH+GHRP6 requieren la integridad del eje hipotálamo-hipofisario. Por ello, en los primeros 5años tras radioterapia hipotalámica debiera valorarse la realización de test de hipoglucemia insulínica si los demás estímulos fueran normales15,17.
Respecto al punto de corte de GH, es importante recordar que la secreción de GH no es la misma a los 16 que a los 50años y, por ello, no es lógico utilizar siempre el mismo valor. La propuesta de la ESPE es utilizar siempre y para todos los estímulos un punto de corte de 5μg/l. Un valor de GH inferior a 5,1μg/l con insulina equivale a un valor menor de 4,15μg/l con GHRH+arginina (95% de sensibilidad y 92% de especificidad para el déficit de GH adulto)5.
Por tanto, existen diferencias en los puntos de corte respecto a cuándo se trata de AO. En este caso, se considera para el tratamiento la deficiencia severa con un valor de GH inferior a 3μg/l tras el test de hipoglucemia insulínica y el test de glucagón18. La hipoglucemia sigue siendo el gold standard, pero tiene bastante variabilidad individual. Por otra parte, no valora diferencias según el IMC, y los pacientes obesos tienen menor respuesta de GH. La falta de punto de corte por IMC tiene implicaciones clínicas. Además, en los estudios publicados19,20, el grupo control incluyó también a sujetos obesos pero sin diferenciar respuestas según IMC. Por otro lado, en la obesidad simple puede detectarse IGF-1 normal e incluso elevado a pesar de existir déficit de GH, ya que puede aumentar la sensibilidad (mayor respuesta de IGF-1 a dosis bajas de GH).
Si se utilizan los criterios de la ESPE en la respuesta a los test de estímulo de GH, tenemos 3 categorías de diagnóstico8:
- -
Déficit de GH en la infancia: pico de GH inferior a 10μg/l.
- -
Déficit de GH en la transición: pico de GH inferior a 5μg/l.
- -
Déficit de GH en el adulto: pico de GH inferior a 3μg/l.
El punto de corte de GH inferior a 5μg/l no está absolutamente aceptado por la administración sanitaria. Esto está cambiando en algunos comités asesores para el tratamiento con GH, donde se están revisando los protocolos y ya se contempla este punto de corte para la transición13. Por otra parte, algunos autores consideran el valor de GH inferior a 6,1μg/l16. Entendemos que la diferencia podría residir en el método de laboratorio empleado para la determinación de GH. Por eso, es importante conocerlo, teniendo en cuenta que en la mayoría de publicaciones los puntos de corte utilizados se definen para métodos de radioinmunoanálisis (RIA).
El test de GHRH+arginina está validado9,21. Tiene la ventaja de su escasa variabilidad individual. Es al menos tan sensible y tan útil como el de hipoglucemia insulínica para reevaluar el déficit de GH en la transición. Solo está contraindicado en la insuficiencia renal, con muy buen perfil de seguridad. No existen diferencias en sexo y edad, pero si en IMC. Así, se considera déficit de GH: para IMC<25kg/m2, GH<11μg/l; IMC 25-30, GH<8μg/l, e IMC>40, GH<4μg/l.
Cuando se utiliza el test de GHRH+GHRP6, se considera déficit de GH cuando el valor es inferior a 10μg/l, aunque faltan estudios amplios y con criterios homogéneos. Si estuviera entre más de 10μg/l y menos de 20μg/l, debiera realizarse otro test.
La utilización de GHRH puede producir efectos secundarios: flushing, vasodilatación, parestesias, náuseas o alteración del sabor. Además, no está disponible en muchos centros.
¿Deben tratarse todos los pacientes deficitarios?Hasta ahora hemos hablado de parámetros bioquímicos, pero no debemos olvidar que el diagnóstico final debiera establecerse considerando pico GH, IGF-1, síntomas, genética (si procede) e imagen en resonancia magnética (RM).
En el consenso de la ESPE de 2005 se defendió ofrecer tratamiento a todos los pacientes deficitarios, mientras que otros autores, más críticos, sostienen que la decisión no debiera basarse únicamente en un punto de corte bioquímico y que debiera realizarse una valoración integral del paciente teniendo en cuenta sus preferencias tras exponer las potenciales ventajas del tratamiento en la edad adulta. En el caso de que el paciente rechazase el tratamiento, debiera seguirse a largo plazo5.
En los casos de IGHD de la infancia que se confirman en la reevaluación, el tratamiento no está autorizado por algunos comités asesores. Sugerimos que se solicite tratamiento a los comités correspondientes y, en los casos en los que no se aceptase, debiera considerarse la posibilidad de enviar de nuevo para valorar tratamiento como «uso fuera de indicación».
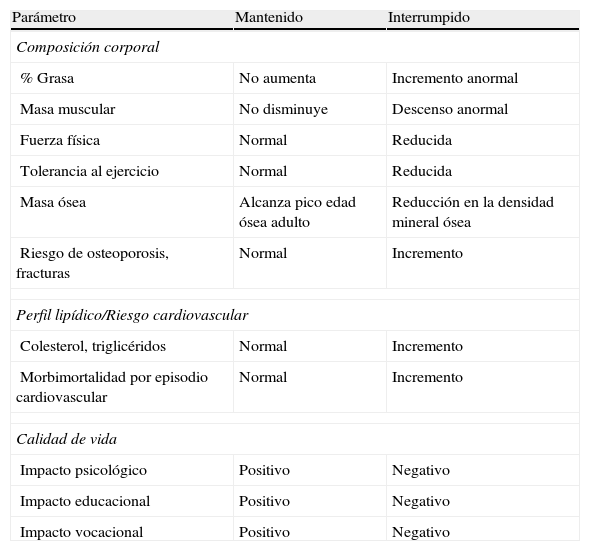
En la tabla 1 se recogen las diferencias a nivel de composición corporal, masa ósea, perfil lipídico y de calidad de vida entre los pacientes que interrumpen o mantienen el tratamiento con GH tras alcanzar la talla adulta1. En la tabla 2 se muestran los efectos de la sustitución con GH en la composición corporal y en el hueso2–5,15.
Riesgos frente a beneficios del tratamiento con GH durante la transición
| Parámetro | Mantenido | Interrumpido |
| Composición corporal | ||
| % Grasa | No aumenta | Incremento anormal |
| Masa muscular | No disminuye | Descenso anormal |
| Fuerza física | Normal | Reducida |
| Tolerancia al ejercicio | Normal | Reducida |
| Masa ósea | Alcanza pico edad ósea adulto | Reducción en la densidad mineral ósea |
| Riesgo de osteoporosis, fracturas | Normal | Incremento |
| Perfil lipídico/Riesgo cardiovascular | ||
| Colesterol, triglicéridos | Normal | Incremento |
| Morbimortalidad por episodio cardiovascular | Normal | Incremento |
| Calidad de vida | ||
| Impacto psicológico | Positivo | Negativo |
| Impacto educacional | Positivo | Negativo |
| Impacto vocacional | Positivo | Negativo |
De Argente et al.1.
Efectos de la sustitución de GH en el periodo de transición
| Composición corporalAumento de masa muscularDisminución de masa grasaIncremento de masa óseaLas mujeres ganan una pequeña cantidad de masa muscular sin cambios en la masa grasaLos hombres ganan bastante masa muscular y pierden una cantidad significativa de masa grasaEl cambio en la relación masa ósea/talla es mayor en los hombresEn el huesoEn déficit de GH al inicio de la infancia hay una reducción significativa del grosor cortical y de la densidad mineral óseaLa suspensión de la GH en la transición limita la adquisición del pico de masa ósea y promueve el desarrollo de osteopenia en la edad adultaEl tratamiento con GH aumenta el contenido mineral óseo en los casos de déficit más grave de GHMetabólicosEl déficit de GH en la transición produce aumento de colesterol total, colesterol LDL y apoproteína B con disminución de HDL. Con la sustitución los cambios son variables según los autores: sin cambios en LDL, colesterol HDL y TG, o bien aumento de HDL o disminución de LDLEl tratamiento con GH disminuye la sensibilidad a la insulina (clamp euglucémico) a pesar de la mejoría simultánea de la composición corporalCalidad de vidaEl único dato con alguna capacidad predictiva es la calidad de vida basal. El sexo, la edad, la intensidad del déficit, otros déficits hormonales asociados, los cambios de composición corporal o los niveles de IGF-1 no tienen ningún valor predictivo |
Otro punto de conflicto es la conveniencia o no del «período de vacaciones», ya que además no existen datos sobre el efecto de estos periodos cortos de interrupción en el estado metabólico de los pacientes durante la transición.
Además hay que tener en cuenta que en otros países, donde el tratamiento no está financiado, la falta de resultados inmediatos fácilmente objetivables (ya no estamos midiendo una talla) puede dificultar la continuidad del tratamiento.
Documentación clínicaClásicamente los tratamientos con GH requieren la aprobación de los comités asesores de tratamiento con GH y sustancias relacionadas. Estos, en la mayoría de los casos son autonómicos, tras la transferencia de las competencias en sanidad a las distintas comunidades autónomas. Los protocolos a cumplimentar debieran incluir la transición considerando los puntos de corte de este período.
Régimen de tratamiento con GHLa dosificación inicial ha pasado de calcularse por el peso, posteriormente por peso ideal y finalmente por aproximación a la dosis del adulto.
Tras alcanzar la talla final no se debe usar la dosis pediátrica de 25μg/kg/día, sino utilizar la de aproximación a la del adulto, manteniendo la IGF-1 normal (entre 0 y +2DE), comenzando con 0,2 a 0,5mg/día. En la transición rara vez se precisan más de 2mg diarios.
En las recomendaciones de la ESPE se aconseja iniciar con 0,2mg/día en varones y 0,3mg/día en mujeres8. Las mujeres sanas segregan 3 veces más GH que los hombres, aunque presentan generalmente niveles de IGF-I séricos similares. Frente a una deficiencia de GH en adultos, la dosis de GH exógena requerida para alcanzar un IGF-I sérico dado es significativamente mayor en mujeres que en hombres (por la resistencia relativa de la GH, en parte mediada por estrógenos).
Monitorización del tratamientoSe deben recordar las interferencias con otros tratamientos hormonales y no olvidar que el cambio de dosis de GH puede hacer necesario el ajuste de las dosis de los restantes tratamientos sustitutivos. La sustitución con estrógenos, una vez completa la pubertad, no debería realizarse por vía oral, ya que por esta vía disminuyen la acción de la GH, con mayor requerimiento de dosis. Además, la GH aumenta la conversión de T4 a T3, por lo que al inicio del tratamiento puede ser necesario el ajuste de levotiroxina. También puede ser necesario aumentar la dosis de hidrocortisona por la acción de la GH sobre la 11-beta-hidroxiesteroidedeshidrogenasa tipo1. Es importante tener en cuenta que el tratamiento de novo puede desenmascarar un hipotiroidismo secundario o una insuficiencia suprarrenal secundaria.
Algún protocolo introduce la realización de ecocardiografía, en base a la posible mejoría de la contractilidad cardiaca tras el inicio del tratamiento demostrada en algunos estudios; sin embargo, las recomendaciones de las guías es que esta solo debiera realizarse si estuviera indicada clínicamente14,15
Seguridad del tratamientoExisten evidencias de que el tratamiento con GH es un tratamiento seguro sin inducir crecimiento tumoral, ni aparición de nuevas neoplasias, aunque estaría contraindicado en pacientes con neoplasias activas15. No existe evidencia de mayor tasa de crecimiento de restos o recidivas en pacientes con tumores hipofisarios o craneofaringiomas. No aumenta tampoco el riesgo de diabetes mellitus tipo1 o tipo215.
Conclusiones y recomendacionesLa transición adecuada de adolescentes con déficit de GH de los servicios de endocrinología pediátrica a los de endocrinología de adultos constituye todo un reto.
En cualquier caso, cada paciente debiera considerarse de forma individualizada aunque se sigan las directrices publicadas.
A continuación reflejamos las recomendaciones que consideramos deben tenerse en cuenta:
- 1.
Recomendamos el tratamiento con GH durante el período de transición, ya que para un déficit similar de GH se han observado diferencias importantes entre CO y AO que pueden deberse a la existencia de períodos sin tratamiento en un momento crucial para la adquisición de una adecuada composición corporal.
- 2.
Recomendamos una colaboración estrecha entre los servicios de endocrinología pediátrica y de adultos con objeto de evaluar de forma conjunta a estos pacientes.
- 3.
Se deben reevaluar todos los casos de déficit de GH, a excepción de las indicaciones pediátricas de tratamiento con GH en niños no deficitarios de GH.
- 4.
La reevaluación debiera realizarse al final del periodo de crecimiento longitudinal.
- 5.
Para la reevaluación, el intervalo sin tratamiento no debiera ser inferior a un mes y el resto de los déficits hormonales deben estar corregidos.
- 6.
Los pacientes con patología orgánica o que presentan alteraciones en la línea media con MPHD o los que presentan IGHD con mutaciones en genes que influyen en el desarrollo de la hipófisis o en la expresión del gen de la GH no precisan reevaluación y el tratamiento podría mantenerse ajustando dosis.
- 7.
Los pacientes con MPHD idiopático o adquirido e IGHD adquirido de etiología desconocida o bien con antecedentes de tumor, cirugía o irradiación hipofisaria, deben reevaluarse tras un mes sin tratamiento, y si la IGF-1 fuera normal, realizar un test de estímulo.
- 8.
Los casos con IGHD con hipófisis normal y sin antecedentes de interés deben reevaluarse tras al menos un mes sin tratamiento y realizar en todos los casos test de estímulo.
- 9.
Por el momento recomendamos la realización del test de hipoglucemia insulínica, ya que sigue considerándose el método de referencia. En los casos en que estuviera contraindicada, podría utilizarse el test de glucagón.
- 10.
Los test de GHRH+arginina o GHRH+GHRP6 requieren la integridad del eje hipotálamo-hipofisario. El test de GHRH+arginina tiene la ventaja de su escasa variabilidad individual y que tiene diferente punto de corte según el IMC.
- 11.
Durante la transición debiera considerarse un punto de corte de GH inferior a 5,1μg/l con insulina, que equivale a un valor menor de 4,15μg/l con GHRH+arginina.
- 12.
La determinación de IGF-1 puede ayudarnos. Hay que tener en cuenta, que tras la suspensión del tratamiento con GH, puede tardar en alcanzar su nivel «basal». El punto de corte de IGF-1 se establece en menos de –2DE (aproximadamente 100μg/dl)2. Se deben recordar las causas de niveles falsamente bajos.
- 13.
La dosis inicial debiera ser de 0,2 a 0,5mg/día. Se deben recordar las interferencias con otros tratamientos hormonales y no olvidar que el cambio de dosis de GH puede hacer necesario el ajuste de las dosis de los restantes tratamientos sustitutivos.
- 14.
Debiera determinarse IGF-1 a las 6semanas del inicio del tratamiento para ajustarlo con objeto de mantener IGF-1 en el percentil 25 a percentil 75 del rango normal. Una vez ajustado, debiera determinarse IGF-1 cada 6meses. Para evaluar cualquier cambio de dosis debería medirse IGF-1 al menos 6semanas después. Anualmente se evaluará peso, IMC, cociente cintura/cadera, tensión arterial, pulso y test de calidad de vida. Cada 2 a 5años se realizará perfil lipídico y densitometría, valorando el T-score en la década de los 20años, y T y Z score posteriormente.
Como en cualquier otra etapa, la evaluación debe realizarse cuando existan datos clínicos que sugieran la presencia de hipopituitarismo. También en aquellos casos en los que a pesar de que no existan síntomas característicos haya antecedentes de procesos que puedan producir afectación del eje hipotálamo-hipofisario, bien sean tumores, procesos inflamatorios o infecciosos, trastornos vasculares, traumatismos, irradiación, alteraciones autoinmunes y genéticas, u otros.
La reevaluación del resto de los ejes hipotálamo-hipofisarios durante el periodo de transición en pacientes que habían iniciado o no el tratamiento en la infancia no ha sido objeto de tantos estudios como en el caso del déficit de GH. No obstante, es importante recordar que en pacientes con hipopituitarismo debe realizarse también la reevaluación del resto de los ejes y definir el momento del paso a endocrinólogos con experiencia en el manejo de la enfermedad hipotálamo-hipofisario en adultos jóvenes9.
Déficit de corticotropina. Insuficiencia adrenal secundaria o centralEn el caso de los niños con hipopituitarismo la valoración del eje hipotálamo-hipofiso-adrenal resulta crucial para la supervivencia a largo plazo. En ocasiones los resultados de la evaluación inicial pueden ser normales, con disminución o pérdida de la función años más tarde10. Aproximadamente un tercio de los que presentan una respuesta normal en la evaluación inicial desarrollan insuficiencia adrenal al final de la pubertad22.
Para evaluar la secreción de ACTH debe medirse el cortisol plasmático entre las 8 y las 9de la mañana. Los resultados deben valorarse siguiendo los criterios habituales para la valoración del eje adrenal.
El test de hipoglucemia insulínica utilizado concomitantemente para valorar GH, y el test de CRH (hormona hipotalámica liberadora de ACTH) son las mejores pruebas para valorar la presencia de insuficiencia adrenal central. El test de glucagón también puede resultar útil en niños. El test de metirapona, tan útil como el de hipoglucemia insulínica, está en desuso por falta de disponibilididad9. El test de ACTH con 1μg (dosis baja) resultó superior al estándar con 250μg para el diagnóstico de la insuficiencia adrenal secundaria en el metaanálisis de Kazlauskaite et al.23. Sin embargo, puede presentar limitaciones. Por una parte, no ha sido validado en pacientes con enfermedades agudas, con trastornos en los ciclos sueño-vigilia o con alteraciones hipotálamo-hipofisarias agudas, como ocurre un mes después de la cirugía. Tampoco ha sido validado el efecto de la comida o de la ingesta de líquido. Además, el valor puede reducirse cuando el test se realiza por la tarde. Por otra parte, no existe información en pacientes con niveles bajos de proteínas, ya que el cortisol circulante se une con alta afinidad a las proteínas. Además, existen problemas de preparación que pueden dar lugar a falsos positivos (niveles de cortisol falsamente bajos a los 30min)23. Para evitarlos, debe seguirse un escrupuloso método de reconstitución y pasos adicionales para evitar la adherencia del preparado24. Por otra parte, es importante tener en cuenta que el tratamiento con GH provoca efectos variables en la respuesta de cortisol a ACTH. Por tanto, el período de interrupción del tratamiento con GH para la reevaluación podría ser un buen momento para revisar la función del eje hipotálamo-hipofiso-adrenal25.
Déficit de tirotropina. Hipotiroidismo secundario o centralPuede deberse a la deficiencia de producción de TSH o de la hormona liberadora de tirotropina. Se diagnostica cuando los niveles de T4 libre están disminuidos con TSH baja o normal26. La TSH basal, que resulta útil para el diagnóstico del hipotiroidismo primario, no permite descartar la presencia de hipotiroidismo central. Clásicamente, para el diagnóstico se utilizaba la determinación de TSH tras estímulo con TRH. Además, este test permitía diferenciar entre el hipotiroidismo central de origen hipofisario, con ausencia de respuesta, del de origen hipotalámico en el que se produce un aumento tardío de los niveles de TSH. Sin embargo, la baja disponibilidad actual de TRH hace necesario la presencia de otros métodos diagnósticos9. Una posibilidad en niños sería observar el pico de TSH nocturna (con un incremento medio del 114% en condiciones normales), asociado a unos niveles de T4 libre en el tercio inferior del rango normal, o incluso menor, que puede ser más sensible que el test de TRH9,26.
Se sugirió que en niños con deficiencia de GH con niveles ligeramente bajos de T4 libre el tratamiento sustitutivo con GH pudiera provocar hipotiroidismo central. Sin embargo, en estudios realizados con determinación del pico nocturno de TSH no se apreciaron alteraciones de la función tiroidea con el tratamiento con GH, salvo que existiera ya un hipotiroidismo central antes del tratamiento26.
Durante la transición, en los pacientes en los no existiera una enfermedad estructural hipotálamo-hipofisaria o no hubieran otras deficiencias además de la de GH y TSH, podría resultar útil reevaluar los niveles de T4 libre, T3 y TSH tras 6 a 12semanas de la retirada del tratamiento. Si los resultados indicaran que existe una función tiroidea normal y se fuera a reanudar el tratamiento con GH, debiera realizarse un seguimiento estrecho de los niveles de T4 libre, reanudando el tratamiento si se observaran niveles diagnósticos de hipotiroidismo central9.
Déficit de gonadotropinas. Hipogonadismo hipogonadotropoLa pubertad retrasada se define como el fallo del inicio del desarrollo mamario y de la aparición del vello púbico a los 13años en la niña, y del crecimiento del testículo y la aparición de vello púbico a los 13años en el niño. Es importante tener en cuenta que esta definición solo es válida si la edad cronológica coincide con la edad ósea. Sin embargo, en pacientes en los que existe déficit de GH la edad ósea puede no coincidir con la edad cronológica y la edad de definición puede retrasarse, y en algunos casos el diagnóstico ocurre precisamente en el período de transición9.
En general, puede asumirse que la pubertad espontánea es poco probable si: a)no existen signos puberales cuando existe una edad ósea de 13años en la niña y 14 en el niño; b)siguen existiendo concentraciones prepuberales de hormona luteinizante y hormona folículo-estimulante medidos por métodos ultrasensibles; c)continúa habiendo niveles prepuberales de estradiol en la niña y de testosterona en el niño, y d)el tamaño gonadal sigue siendo prepuberal. Para valorar este dato se requiere estudio ecográfico en las niñas. En tales casos, el comienzo del tratamiento hormonal sustitutivo vendrá definido por la edad cronológica, la madurez emocional, la talla potencial y la densidad ósea9.
Tratamiento del déficit del resto de las hormonas de la hipófisis anterior durante la transiciónDéficit de corticotropina. Insuficiencia adrenal secundaria o centralEs el primer déficit que debe ser sustituido —y además de forma urgente— ante su sospecha, ya que puede comprometer la vida del paciente. La administración de una dosis correcta de esteroides en la etapa de transición resulta crucial. Requiere por una parte evitar la utilización de dosis subóptimas que podrían provocar hipoglucemia o incluso crisis adrenal27, y por otra la sobredosificación que podría provocar detención del crecimiento, obesidad, debilidad muscular y osteopenia28.
El glucocorticoide más utilizado es la hidrocortisona (Hidroaltesona®, en comprimidos de 20mg). Es un preparado de vida media corta que, distribuido en 2 o 3dosis diarias, permite imitar el ritmo circadiano del cortisol. Preparados de vida media más prolongada, como la prednisona, la prednisolona o la dexametasona, son menos utilizados.
La dosis a emplear es la equivalente a la producción diaria de cortisol, entre 12 y 14mg/m2 y día, habitualmente de 15 a 20mg/día29,30. En la etapa de la transición se recomienda su administración dividida en 3dosis diarias para evitar el pico que se produce tras la dosis de la mañana y los niveles bajos durante la noche, habituales en los regímenes de 2dosis al día31,32. Plenadren® es una hidrocortisona de liberación modificada que consigue imitar el ritmo fisiológico de secreción del cortisol en una administración única diaria. No está comercializada en España, aunque sí disponible su utilización como medicación extranjera. Sin embargo, actualmente su indicación está restringida a la insuficiencia adrenal en adultos, y no hay datos sobre su uso en la etapa de transición.
En caso de estrés leve y moderado por proceso intercurrente (fiebre, infecciones, cirugía menor), el paciente debe duplicar la dosis habitual (24mg/m2 y día). Ante cualquier situación que impida el tratamiento por vía oral, será necesario recurrir a la administración de hidrocortisona por vía intramuscular o intravenosa (Actocortina® 50-100mg por vía parenteral y repetir a las 8-12h en función de la evolución clínica).
En casos de estrés mayor, como la cirugía o quemados, se debe emplear la vía parenteral para la administración de la hidrocortisona, siendo la pauta habitual 100mg de Actocortina® cada 6-8h.
La monitorización del tratamiento se realiza con datos clínicos y bioquímicos, ya que no existe ningún marcador hormonal que nos permita evaluar si la dosis administrada es la adecuada.
Déficit de tirotropina. Hipotiroidismo secundario o centralEl tratamiento del déficit de TSH se realiza con levotiroxina por vía oral, en una sola toma diaria y de preferencia por la mañana, en ayunas. Es importante recordar que nunca debería iniciarse el tratamiento con levotiroxina hasta descartar la existencia de un déficit de ACTH, pues podría desencadenar una crisis adrenal.
El tratamiento es esencial para la maduración y el desarrollo intelectual del paciente, aunque, al igual que ocurre con el déficit de ACTH, debe evitarse la sobredosificación para no comprometer el crecimiento y condicionar la talla final.
La dosis total por kilogramo de peso en la etapa de transición es de aproximadamente 2μg/kg/día, menor que los requerimientos en la infancia pero mayor que los habituales en la edad adulta. La monitorización del tratamiento se realiza con la determinación de T4 libre, aumentándose de forma progresiva la dosis cada 8-10 semanas hasta mantener el nivel de T4 libre en el punto medio del rango normal.
Déficit de gonadotropinas. Hipogonadismo hipogonadotropoMujeresEl objetivo del tratamiento es lograr un desarrollo puberal normal, y debe iniciarse en torno a los 12años para inducir la pubertad.
El tratamiento se realiza con estrógenos. Disponemos de 2formas de administración oral (etinilestradiol y valerato de estradiol) y una de administración transdérmica (parches de estradiol).
El tratamiento se iniciará con dosis muy bajas, entre un octavo y un décimo de la pauta habitual en la edad adulta, para favorecer la ganancia de masa ósea y no comprometer el crecimiento longitudinal33. Para el inicio y sobre todo en la inducción de los caracteres sexuales secundarios el etinilestradiol es el estrógeno más recomendado34. La dosis de inicio es de 2,5-5μg/día con el etinilestradiol33 o parches de 25μg/día de estradiol.
Posteriormente, la dosis se dobla cada 6meses durante los 3años siguientes hasta alcanzar la dosis utilizada en mujeres adultas (20-35μg/día de etinilestradiol, 2-4mg de estradiol oral o 50 a 100μg/día de estradiol transdérmico35).
Tras completar el desarrollo mamario y/o tras la primera menstruación es preciso asociar progestágenos de forma cíclica los últimos 12días del ciclo para conseguir hemorragia por deprivación y prevenir el cáncer de endometrio.
Para el mantenimiento, la vía transdérmica es preferible frente a los preparados por vía oral, ya que estos últimos se han relacionado con un empeoramiento del perfil metabólico y la composición corporal. Además, bloquean la síntesis hepática de IGF-1, factor a tener en cuenta en las pacientes que reciban tratamiento sustitutivo con GH. Cuando se emplee la vía oral debiera utilizarse la dosis mínima de estrógenos posible36,37.
Cuando se desea inducir la ovulación, el tratamiento se realiza con gonadotropinas como FSH recombinante (75-150UI diarias) hasta que los folículos presenten un tamaño de 16-18mm o el nivel de estrógenos sea mayor o igual a 200pg/ml, momento en el que se administrará hCG (5.000UI) para inducir la ovulación en 36h. La monitorización ecográfica es fundamental. El tratamiento con GnRH pulsátil solo es útil cuando la capacidad secretora de FSH y LH está conservada, pero no aporta ventajas.
HombresLa sustitución se realiza con derivados de la testosterona. Los preparados disponibles son de administración parenteral o transdérmica.
Por vía transdérmica contamos con parches (presentaciones de 15 a 30mg) y geles (presentaciones de 25 y 50mg). Ambos son de aplicación diaria y mantienen niveles normales y estables de testosterona durante 24h.
Dentro de los preparados para la administración parenteral disponemos de enantato y cipionato de testosterona (presentaciones de 100 y 250mg), que requieren administración cada 14 a 28días, y de undecanato de testosterona (presentación de 1.000mg). Este último es un preparado de vida media larga que proporciona niveles estables de testosterona a lo largo de 12semanas.
En la adolescencia son de elección el enantato o cipionato de testosterona por vía parenteral. La dosis de inicio es de 25 a 50mg cada 4semanas (vía intramuscular) y se incrementa progresivamente cada 6 a 12meses hasta 250mg cada 2 a 4semanas33. Esta pauta ha demostrado su eficacia en la inducción de los caracteres sexuales secundarios y favorece el crecimiento lineal sin comprometer la maduración ósea. El valor de testosterona permite la monitorización del tratamiento, precisando estar próxima a la mitad del rango normal. Si se utilizan parches o geles, la dosis es de 5 y 50mg/día, respectivamente.
Clásicamente, se ha utilizado la HCG (500-1.000UI, 2 a 3veces por semana) para inducir fertilidad, o en niños para aumentar el volumen testicular. Si no existe respuesta adecuada tras 12semanas de tratamiento puede añadirse FSH recombinante (75-100UI, 3veces por semana). Sin embargo, no existe evidencia disponible de que dicho tratamiento restaure o mejore la espermatogénesis. La administración de pulsos de GnRH no presenta ventajas frente al empleo de gonadotropinas, y su coste económico es mayor.
Conflicto de interesesE. Fernández y J. M. Recio no tienen conflictos de intereses. I. Bernabeu declara haber recibido honorarios como ponente de Pfizer e Ipsen. C. Fajardo y C. Álvarez Escolá declaran haber recibido honorarios por participar como ponentes o moderadoras en reuniones organizadas por Pfizer, Lilly, Novo Nordisk e Ipsen.
Queremos expresar nuestro agradecimiento a los miembros del Área de Conocimiento de Neuroendocrinología de la SEEN, que revisaron este manuscrito. También al Dr. Elías Delgado, Vicepresidente de la SEEN y Coordinador de Áreas de conocimiento y Grupos de Trabajo, por las modificaciones sugeridas y la concesión del aval de la Junta Directiva de la SEEN para su publicación.
