


Partial androgen insensitivity syndrome (PAIS) is a 46,XY disorder of sexual differentiation where there is a loss of functions of androgen receptors (ARs). PAIS is a condition caused by mutation in the AR (Xq11–12) gene that encodes for AR.
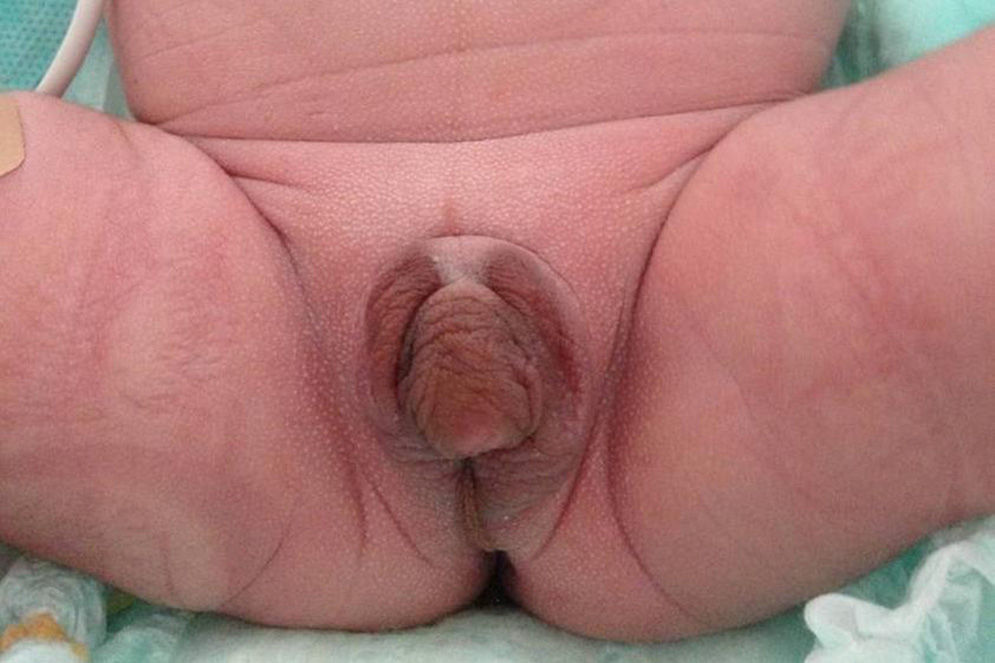
We report the case of a term newborn from a controlled, uncomplicated pregnancy. He was the second child of a father of Italian origin and a Dominican mother who were healthy and not related. Prenatal ultrasound examinations were normal and suggested a female infant. Parameters recorded at birth included: weight, 2862g (−0.12SD); length, 46.5cm (−0.62SD); head perimeter, 35cm (1.16SD); and Apgar score, 8–9. The infant had hyperpigmented, ambiguous genitalia, with a 1.5cm genital tubercle with hypospadic urinary meatus, rims of symmetrical scrotal labia, fused in their posterior portion, and no vaginal opening or testes on palpation, corresponding to Prader 4–5 (Fig. 1). The infant had mild respiratory distress secondary to pulmonary hypertension with nonobstructive hypertrophic cardiomyopathy which resolved in two weeks. Acid–base balance and sodium and potassium levels were normal. Abdominal ultrasound performed on the first day of life showed a structure consistent with uterus and a fluid-filled endometrial cavity. No ovaries or testes were seen.

The results of hormonal tests performed in the first week of life included: LH<0.1IU/mL; FSH, 0.4mIU/mL; testosterone, 152ng/dL; cortisol, 7μg/mL; ACTH, 23.3pg/mL; androstenedione, 5.5ng/mL; DHEAS, 159μg/mL; aldosterone, 1428pg/mL; 17-hydroxyprogesterone, 9ng/mL; renin, 12.2ng/mL/h; and 17-beta estradiol<12pg/mL; dihydrotestosterone, 28.2ng/dL; they all were within the normal values. The results of subsequent tests included 11-deoxycortisol, 6.15ng/mL; deoxycorticosterone 3ng/mL; inhibin B, 99pg/mL; and anti-Müllerian hormone, 18.8ng/mL, also within the normal levels for age.
Karyotype was 46,XY, consistent with a 46,XY disorder of sexual differentiation with ambiguous genitalia, and a study for microdeletion in the SRY gene was normal. Analyses were requested of the androgen receptor gene and SRD5A2 gene, associated with 5-alpha-reductase deficiency, because the mother was born in the region of the Dominican Republic where this syndrome was reported.
Serial voiding cystourethrography showed a urethra of male morphology with urogenital sinus, and repeat ultrasonography at 10 days of life revealed the presence of paravesical structures consistent with gonads with no follicles inside, 0.8cm and 1cm in size. Based on the finding of 46,XY genetic sex, the production of normal testosterone levels by a structure different from the adrenal gland (in a probable testicular tissue, possibly in the gonad), the urethra with the length and course of male characteristics, and a penis of acceptable size, and after agreement with the departments of pediatric endocrinology and surgery and neonatology, and the family, the infant was assigned a male gender at 18 days of life.
At 36 days of life, the infant underwent surgery for left inguinal-scrotal hernia, consisting of orchiopexy and gonadal biopsy. A testicular bìopsy showed marked germ cell hypoplasia (type IIof Nistal et al.1) and gonadal 46,XY karyotype.
When faced with a newborn with ambiguous genitalia and a structure consistent with an uterus in emergency ultrasonography, the most likely diagnosis is virilization of a 46,XX female, and the most common cause is congenital adrenal hyperplasia due to 21-hydroxylase deficiency. When a 46,XY genetic sex is shown, the 46,XY disorder of sexual differentiation is more complex to characterize.2 Analysis of the SRY gene, the main gene involved in primary gonadal differentiation, was normal. Vagina and uterus remnants could correspond to a persistent Müllerian duct syndrome due to deficient anti-Müllerian hormone production or receptor deficiency. Undervirilization could in turn be due to PAIS, although this is not usually associated with Müllerian remnants. 5-alpha-reductase deficiency could partly explain the condition, and the mother came from Las Salinas,3 the area in the Dominican Republic where this disease was discovered, but where there is also a high rate of ovotesticular chimera and mixed gonadal dysgenesia. Other associated symptoms which involve kidney disease, brain changes, or cardiac hypertrophy, such as the LEOPARD syndrome, are unlikely, but should be evaluated based on findings.
Molecular study of the AR gene detected at two months the c.1139C>G mutation with the substitution of proline at position 380 for arginine, associated with PAIS, which confirmed the diagnosis. This mutation has only been reported in one other patient (Audi et al.)4 from Gambia with PAIS and Müllerian remnants. Study of the SRD5A2 gene showed a change in homozygosis at position 265 (c.265C>G), with the substitution of leucine for valine (p.Leu89Val), of uncertain significance.5
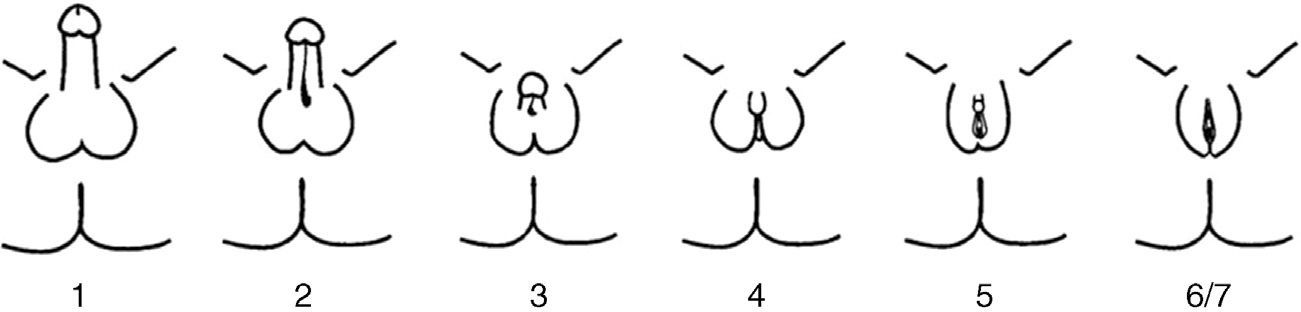
PAIS is an X-linked recessive disease caused by mutations in the AR gene. This gene is located in the Xq11–12 region and encodes for AR.6 AR mutations only occur in 20% of patients with PAIS. PAIS prevalence is unknown because of the variability of its clinical manifestations and the existence of atypical forms. It has been estimated that PAIS may be 10 times less common than complete androgen insensitivity syndrome (CAIS). PAIS occurs in 46,XY individuals with a highly variable clinical spectrum, from almost complete feminization to almost normal masculinization, through individuals with frank sexual ambiguity. Clinical variability reflects the different types of mutations that may affect the AR gene. The predominantly male phenotype exhibits micropenis, variable hypospadias, and cryptorchidism. During puberty, eunuchoid appearance and breast development (gynecomastia) are seen. The predominantly female phenotype shows clitoromegaly, fused labia, and pubic hair during puberty. The more ambiguous forms are discovered at birth or during infancy. The most adequate phenotypic classification is not the Prader classification, but the one by Quigley et al.7 (Fig. 2). The internal genitalia, derived from the Wolffian duct, may be partially or totally developed, while testes are most often undescended (in the inguinal region or scrotum/labia majora), have few or no germ cells, and are more susceptible to malignization in adulthood. Although it has been reported that there should be no Müllerian remnants, there have been individual reports of the persistence of these structures in patients with CAIS8–10 or PAIS, as occurred in our patient; the mechanism of this association has not been clearly elucidated yet. Prognosis depends on the degree of genital ambiguity and on physical and psychosocial adaptation to the assigned sex. Complications may occur in adult age, with a greater trend to gonad malignancy, infertility, and osteoporosis.

When ambiguous genitalia exist, gender assignment is an urgent but complex process that requires evaluation by a multidisciplinary team encompassing the family, neonatologists, radiologists, pediatric surgeons, endocrinologists, and psychologists. Gender should be assigned as soon as possible, but only when adequate information is available, in the best interest of the newborn.11
Please cite this article as: Bermejo-Costa F, Lloreda-García JM, Donate-Legaz JM. Síndrome de insensibilidad parcial a andrógenos con restos müllerianos. Descripción de un caso. Endocrinol Nutr. 2015;62:469–471.
