



The increase in cardiovascular morbidity and mortality associated to insulin resistance (IR) states (obesity, metabolic syndrome, type 2 diabetes) represents a major public health problem. In IR, dyslipidemia typically include hypertriglyceridemia, low high density lipoprotein cholesterol, increased small and dense low density lipoprotein particles, and post-prandial hyperlipidemia, which play a direct or indirect role in the mechanisms of atherosclerosis. Dyslipidemia is mainly due to accumulation of circulating triglyceride-rich lipoproteins from the liver and bowel. The bowel has traditionally been seen as a passive organ, but current evidence confirms that it is an active organ subject to regulation by free fatty acids, insulin, incretins, and inflammation. Two new concepts have emerged: intestinal IR and overproduction of chylomicrons in hyperinsulinemic/IR states. A better understanding of intestinal IR may make the enterocyte a therapeutic target.
El aumento de la morbimortalidad cardiovascular en estados de resistencia insulínica (RI), como la obesidad, el síndrome metabólico, y la diabetes tipo 2, representa un problema mayor para la salud pública. La dislipemia de la RI comprende la hipertrigliceridemia, una disminución de colesterol de las lipoproteínas de alta densidad, aumento de las lipoproteínas de alta densidad pequeñas y densas e hiperlipemia posprandial, cumpliendo un papel directo e indirecto en la arterosclerosis. Esta dislipemia es debida a la acumulación de lipopartículas ricas en triglicéridos de origen intestinal y hepático. El intestino ha sido considerado un órgano pasivo, pero la evidencia actual confirma al intestino como un órgano activo sometido a la regulación de: ácidos grasos libres, insulina, incretinas e inflamación. Dos conceptos han surgido, el de la RI intestinal y el de la sobreproducción de quilomicrones en los estados de hiperinsulinismo/RI. Una comprensión de la RI intestinal puede convertit al enterocito en una diana terapéutica.
The alarming increase in the prevalence of insulin resistance (IR) in conditions such as type 2 diabetes, familial combined hyperlipidemia (FCHL), and metabolic syndrome, combined with the current obesity epidemic, represents a significant public health problem.1 Atherosclerosis and its coronary, cerebral, and peripheral vascular complications represent the leading cause of morbidity and mortality in IR states, with a relative risk of 2–3 in males and 4–5 in females with type 2 diabetes mellitus.2
Lipid disorders play a predominant role in cardiovascular disease associated with IR states. The main lipid risk factor is low density lipoprotein cholesterol (LDL-C). This observation has been supported by epidemiological studies and fully concordant intervention studies.3 Dyslipidemia in IR is characterized by the following four manifestations: fasting hypertriglyceridemia, low levels of high density lipoprotein cholesterol (HDL-C), an increased number of small, dense LDL particles, and postprandial hyperlipidemia.4 Each of these parameters represents an independent cardiovascular risk marker.5–9 There are many arguments supporting the hypothesis that cardiovascular risk does not completely disappear with low LDL-C levels, particularly in conditions such as diabetes. This is now defined as residual cardiovascular risk.10
Some authors attribute part of the atherogenic process to an increased production of triglycerides (TG) in the bowel in the postprandial period.9,11 In fact, postprandial hyperlipidemia has been identified as a vascular risk factor in cohort and case–control studies.12,13 Recent studies suggest that type 2 diabetic patients experience an increase in triglyceride-rich lipoproteins (TRLs) of intestinal origin and in the postprandial state14,15; a significant association has been found between postprandial lipid levels and coronary atherosclerosis.16 Unfailingly, intestinal TRL remnants are able to enter the subendothelial space of the vascular wall and to participate in the atherosclerosis process.17 The purpose of this study was to summarize recent findings referring to the new role of enterocytes in relation to the pathophysiology of dyslipidemia of IR states.
Role of triglyceride-rich lipoproteins in atherogenesisClassical viewIn humans, apolipoprotein (apo) B-48 is synthesized by the small bowel from the mRNA of apoB-100 by a post-transcriptional mechanism called mRNA editing, necessary for chylomicron (CM) synthesis, assembly, and secretion.18–20 There is a single apoB-48 molecule per CM21; it is therefore possible to differentiate in humans TRLs of hepatic origin carrying apoB-100 (TRL-apoB-100) from TRLs of intestinal origin carrying apoB-48 (TRL-apoB-48). ApoB-48 content is determinant of the number of intestinal TRL particles and their remnants, of which the latter have greater atherogenic power. By contrast, TG content is a determinant of particle size; greater size decreases the atherogenic potential of TGs, as they cannot cross the subendothelial space.22
In subjects with IR, dyslipidemia is due to a large extent to the accumulation of hepatic and intestinal TRLs in plasma. This accumulation is also secondary to hepatic hyperproduction of type 1 (big and rich in TG) very low density lipoproteins (VLDL),23 to a defect in TRL clearance which may be attributed to decreased lipoprotein lipase (LPL) activity,24,25 to an abnormal apoprotein composition of lipoproteins,26 to an increased circulating pool of plasma TRLs resulting from competition between hepatic and intestinal TRLs using the same saturable clearance route of LPL,27 and to a defect in hepatic uptake.28
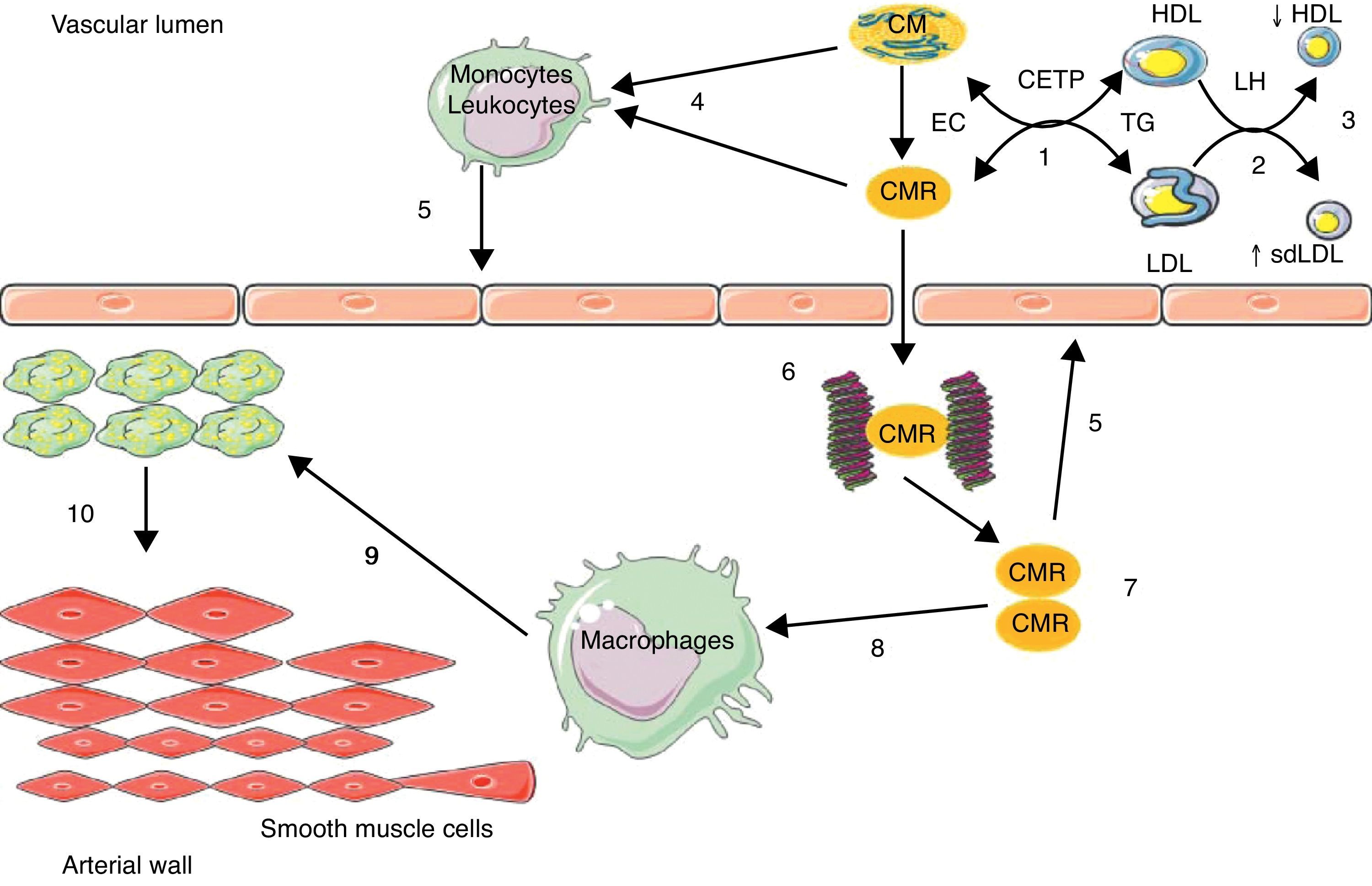
The increased residence time in plasma of TRLs facilitates TG transfer from these particles to LDL and HDL by the action of the cholesteryl ester transfer protein (CETP); LDL and HDL enriched in TG are privileged substrates for hepatic lipase (HL), whose action is responsible for the formation of small, dense LDL and for increased HDL catabolism.23 TRLs act indirectly in atherosclerotic events, promoting the occurrence of two atherogenic lipid abnormalities: an increase in the number of small, dense LDL, and a decrease in HDL-C levels (Fig. 1).
 Enrichment of low density lipoprotein (LDL) and high density lipoprotein (HDL) in triglycerides (TG) through the cholesteryl ester transfer protein (CETP); (2) TG hydrolysis by hepatic lipase (HL); (3) increase in small, dense LDL (sdLDL) and decrease in HDL by hypercatabolism; (4) enrichment of leukocytes/monocytes in free fatty acids; (5) activation of endothelium; (6) binding of chylomicron remnants (CMRs) to proteoglycans; (7) accumulation of CMRs; (8) binding of CMRs to apoB-48 receptors in macrophages; (9) conversion of macrophages into foam cells; (10) migration and proliferation of smooth muscle cells from the media. EC: esterified cholesterol.")
Implication of intestinal triglyceride-rich lipoparticles in atherosclerosis.
(1) Enrichment of low density lipoprotein (LDL) and high density lipoprotein (HDL) in triglycerides (TG) through the cholesteryl ester transfer protein (CETP); (2) TG hydrolysis by hepatic lipase (HL); (3) increase in small, dense LDL (sdLDL) and decrease in HDL by hypercatabolism; (4) enrichment of leukocytes/monocytes in free fatty acids; (5) activation of endothelium; (6) binding of chylomicron remnants (CMRs) to proteoglycans; (7) accumulation of CMRs; (8) binding of CMRs to apoB-48 receptors in macrophages; (9) conversion of macrophages into foam cells; (10) migration and proliferation of smooth muscle cells from the media. EC: esterified cholesterol.
TRLs may act directly in the atherosclerosis process through an enrichment in circulating free fatty acids (FFAs), which are endogenous ligands of toll-like receptors present in leukocytes,29 leading to the activation of nuclear transcription factor Kappa B (NFkB),29 thus generating the synthesis of inflammatory and procoagulant cytokines and maintaining a proinflammatory, procoagulant, and proatherogenic microenvironment in the arterial wall, with its deleterious consequences11 (Fig. 1).
A more direct action involves the retention of remnants of hepatic and intestinal TRLs in the arterial wall. In fact, a binding site of apoB-48 has been identified in proteoglycans in arterial intima.30 Moreover, a specific receptor of apoB-48 was isolated and characterized in macrophages and monocytes.31,32 This receptor (apoB-48-R) differs from LDL receptors apoB/E, LRP (LDL-receptor related protein), and scavenger receptors in that it only captures TRL-apoB-48 from the bowel. This receptor is present at foam cell level in lipid streaks and atheroma plaques. Its capacity to bind and internalize intestinal TRL remnants is likely to be involved in the acquisition of the foam cell phenotype of macrophages and in the formation of atherosclerotic lesions (Fig. 1). ApoB-48 has recently been isolated from aortic atheroma plaques in humans.33 The retention capacity of remnants of TRL-apoB-100 is ten times higher as compared to remnants of TRL-apoB-48 based on plasma levels, but cholesterol levels are 40 times higher in TRL-apoB-48 as compared to TRL-apoB-100. This gives TRL-apoB-48 the possibility of releasing four times more cholesterol in the vascular wall than TRL-apoB-100.17
Based on all of these data, TRL-apoB-48 of dietary origin, CMs and their remnants appear to have atherogenic potential. Data from animal studies support this atherogenic role. In fact, there are transgenic mouse models (ApoCIII+/+ and ApoE−/−) characterized by a marked increase in lipoprotein particles carrying apoB-48 (although apoB-48 is not bowel-specific in mice). These animals experience accelerated atherogenesis, particularly if lipid contents from their diet are increased.34,35
It may be concluded from the foregoing that, apart from LDL (particularly the small, dense subfraction), there are other atherogenic lipid particles: CMs and their remnant of dietary origin, with apoB-48 having not only a role as a CM marker, but also an active role in atherosclerotic plaque formation. The classical view of the accumulation of intestinal TRLs in blood is secondary to decreased CM clearance in IR states.
Exploration of postprandial lipidemiaMetabolism of intestinal TRLs may be analyzed using two methods: the first method is the area under the curve (AUC) of CM markers such as vitamin A and/or apoB-48 after an oral lipid overload (OLO),36 and the second is the apoB-48 kinetic method using an infusion of stable isotopes under steady state conditions (plateau).39
The first method uses a vitamin A metabolite, retinyl palmitate (RP), because it is incorporated into the CM and then recaptured by the liver and not secreted subsequently.37 ApoB-48 is secreted in the bowel only and is not exchangeable with other lipid fractions, which makes it a helpful CM marker.38 The differences lie in the fact that RP is 18–25% exchangeable with LDL and that it is only incorporated into mature CMs and does not integrate into newly formed CMs. These two differences are absent when apoB-48 is used as a specific intestinal marker.36
The kinetic method represents the gold standard for the study of CM metabolism and consists of the administration every 30 or 60min of nutritional products adapted to the daily calorie requirements of each patient to stimulate apoB-48 synthesis, obtaining stable apoB-48 plasma levels39,40 (plateau). In parallel to nutrition, a stable isotope such as the amino acid leucine labeled with deuterium (2H3-leucine) or carbon 13 (1-13C-leucine) is infused in order to be incorporated into newly synthesized apoB-48. The goal is to obtain a ratio between the apoB-48 incorporating the stable isotope and the apoB-48 not incorporating the isotope. This ratio is expressed as a percentage, thus obtaining an isotopic enrichment curve. These values are entered into a multicompartmental mathematic model to calculate the production rate (PR) and the catabolic rate (CR) of apoB-48.41 To sum up, the method estimating the AUC of apoB-48 after OLO is the simplest procedure, but also the least precise; by contrast, the kinetic method, in which the PR and CR of apoB-48 are calculated, is more accurate, although more complex.
Regulation of the synthesis of intestinal triglyceride-rich lipoproteinsModulation of dietThe production of intestinal TRLs has been considered for a long time to be regulated by dietary supply and, more specifically, by the total of lipids ingested. Many studies have addressed the relation between dietary supply and postprandial lipidemia without making any differentiation between abnormal production and/or clearance of CMs and their remnants. Lipid amounts of 20–50g per meal dose-dependently increase postprandial lipidemia, and a constant increase is seen with amounts higher than 80g per meal.42 The addition of sucrose and/or fructose or carbohydrates with a high glycemic index to a fat-rich meal increases postprandial lipidemia.42
The other dietary factors that may play a mild to moderate role in the reduction of postprandial lipidemia include the administration of n-3 (omega-3) and n-6 (omega-6) polyunsaturated fatty acids or fiber-rich foods.42
Increased postprandial lipidemia may also be secondary to a cholesterol-deficient or protein-rich meal.43 Dietary provision contributes to a marked increase in TG content rather than apoB-48 content in the CM particle, with a resultant increase in the size, rather than the number, of TRL particles.22
Overproduction of intestinal triglyceride-rich lipoproteins in insulin resistance statesAnimal modelThe Syrian golden hamster (fructose-fed hamster), made resistant to insulin through fructose-rich feeding, is a useful animal model for studying the role of the production of intestinal TRLs in the development of metabolic dyslipidemia.44 As occurs in humans, the tissue-specific expression of apoB-100 in the liver only and of apoB-48 in the bowel only represents an advantage in the hamster model, allowing for in vivo studies of the production of intestinal versus hepatic TRLs.45,46 In this model, a 2–4-fold greater increase in TRL production was shown as compared to the hamster model with no IR. This acute secretion of intestinal TRLs has been confirmed ex vivo in cultures of enterocytes with IR. Similar to hepatic TRL overproduction,47–49ex vivo and in vitro experiences on enterocytes have shown that chronic fructose feeding is associated with a greater stability of intracellular apoB-48, increased de novo lipogenesis, increased endogenous synthesis of TG and cholesterol esters, and increased expression of microsomal transfer protein (MTF), a key protein in the formation of intestinal lipoproteins44 (Fig. 2). A fructose-rich diet for only two days has no impact on intestinal TRL production, which suggests that this fructose-rich diet has a chronic effect.44

Mechanisms of overproduction of intestinal triglyceride-rich lipoparticles in the setting of hyperinsulinism and insulin resistance.
FFAs: free fatty acids; ω3FAs: omega-3 fatty acids; FAT/CD36: fatty acid translocase; GLP-1: glucagon-like peptide-1; GLP-2: glucagon-like peptide-2; MTP: microsomal transfer protein; SREBP-1c: sterol response element binding protein 1c; TNFα: tumor necrosis factor alpha; TG: triglycerides.
This overproduction of intestinal TRLs has been confirmed in vivo, ex vivo, and in vitro in another IR model, the high fat-fed Syrian Golden hamster.50 In these two animal models of hamster with IR, treatment with rosiglitazone (an agonist of the peroxisome proliferator-activated receptor gamma (PPAR-γ)) corrected elevated MTP levels and reduced the intestinal overproduction of TRLs, probably promoting insulin sensitivity.50,51 The results in this model were very similar to those in the model with IR/type 2 diabetes (the gerbil Psammomys obesus), which supports the association between IR and intestinal TRL overproduction. In this model, increases in monoacylglycerol transferase (MGAT), diacylglycerol transferase (DGAT), and the expression of fatty acid binding protein L (L-FABP) promoted de novo lipogenesis, without finding any abnormalities in MTP expression.52
As regards insulin signaling pathways, in vivo, ex vivo and in vitro experiences in the hamster model with IR (fructose-fed hamster) showed defects at enterocyte level including a decrease in insulin receptor substrate-1 (IRS-1) and an inhibition of protein kinase B or Akt, and inversely, increased concentrations of subunit p110 of PI-3 kinase, tyrosine phosphatase 1-B, and the mitogen-activated protein kinase pathway (MAPK/ERK). These abnormalities suggest intestinal IR, which causes hyperactivation of the MAP/ERK pathway, leading to the increased expression of MTP, apoB, and a transcription factor, sterol response element binding protein 1c (SREBP-1c), which stimulates the synthesis of proteins involved in de novo lipogenesis.53,54 Overall, these abnormalities lead to the increased production and secretion of intestinal TRLs53,54 (Fig. 2).
Human modelThe overproduction of intestinal TRLs is recognized as a new component of IR syndrome. This excess production was first documented in humans, in whom intestinal TRL production is increased as a function of the degree of hyperinsulinism and insulin sensitivity.41 Unlike animal models, rosiglitazone does not improve the production of intestinal TRLs in healthy subjects, but impairs it despite its insulin sensitizing effect, with a non-significant trend to an increased production and decreased clearance of intestinal TRLs55 (Table 1). These results may explain the increase in coronary atherosclerosis in patients treated with rosiglitazone.56 Observational studies in obese patients with IR have shown increased plasma levels of apoB-48 in fasting and postprandial conditions.57,58 In patients with type 2 diabetes, the overproduction of intestinal TRLs was found to be associated with a decrease in their clearance.14 FCHL is characterized by higher apoB-48 levels in fasting and postprandial conditions59,60 (Table 2). It is well known that the accumulation of apoB-48 is caused by a decreased LPL activity secondary to the increase in hepatic TRLs60 associated in turn with apoC-III increase (a physiological inhibitor of LPL). ApoC-III in turn stimulates TRL production, participating in the final stage of TRL assembly at cell level.61 The presence of liver steatosis in type 2 diabetes and in FCHL is associated with increased production of apoB-48 production in the postprandial state.62,63 The IR state is the main determinant of apoB-48 overproduction, with active involvement in the coronary disease of these patients.64
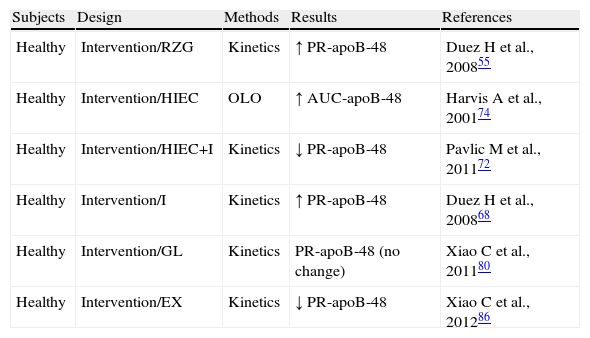
Exploration of the metabolism of triglyceride-rich lipoproteins (TRLs)-apoB-48 in preserved insulin sensitivity states.
| Subjects | Design | Methods | Results | References |
| Healthy | Intervention/RZG | Kinetics | ↑ PR-apoB-48 | Duez H et al., 200855 |
| Healthy | Intervention/HIEC | OLO | ↑ AUC-apoB-48 | Harvis A et al., 200174 |
| Healthy | Intervention/HIEC+I | Kinetics | ↓ PR-apoB-48 | Pavlic M et al., 201172 |
| Healthy | Intervention/I | Kinetics | ↑ PR-apoB-48 | Duez H et al., 200868 |
| Healthy | Intervention/GL | Kinetics | PR-apoB-48 (no change) | Xiao C et al., 201180 |
| Healthy | Intervention/EX | Kinetics | ↓ PR-apoB-48 | Xiao C et al., 201286 |
AUC-apoB-48: area under the curve of apoprotein B48; HIEC: hyperinsulinemic-euglycemic clamp; EX: exenatide; GL: glucagon; I: Intralipid; RZG: rosiglitazone; no change: no significant change; OLO: oral lipid overload; PR-apoB-48: apoprotein B48 production rate.
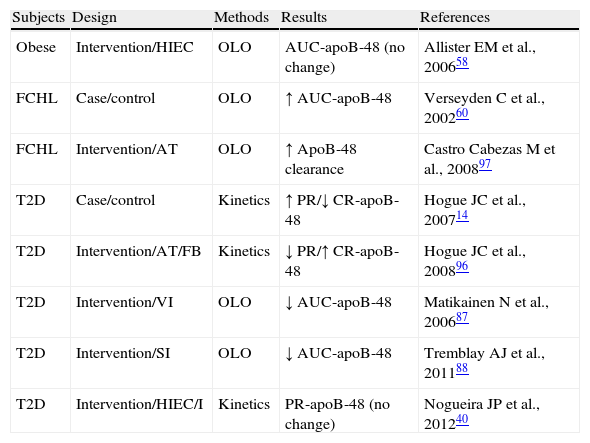
Exploration of the metabolism of triglyceride-rich lipoproteins (TRLs)-apoB-48 in insulin resistant states.
| Subjects | Design | Methods | Results | References |
| Obese | Intervention/HIEC | OLO | AUC-apoB-48 (no change) | Allister EM et al., 200658 |
| FCHL | Case/control | OLO | ↑ AUC-apoB-48 | Verseyden C et al., 200260 |
| FCHL | Intervention/AT | OLO | ↑ ApoB-48 clearance | Castro Cabezas M et al., 200897 |
| T2D | Case/control | Kinetics | ↑ PR/↓ CR-apoB-48 | Hogue JC et al., 200714 |
| T2D | Intervention/AT/FB | Kinetics | ↓ PR/↑ CR-apoB-48 | Hogue JC et al., 200896 |
| T2D | Intervention/VI | OLO | ↓ AUC-apoB-48 | Matikainen N et al., 200687 |
| T2D | Intervention/SI | OLO | ↓ AUC-apoB-48 | Tremblay AJ et al., 201188 |
| T2D | Intervention/HIEC/I | Kinetics | PR-apoB-48 (no change) | Nogueira JP et al., 201240 |
AUC-apoB-48: area under the curve of apoprotein B48; AT: atorvastatin; HIEC: hyperinsulinemic-euglycemic clamp; FB: fenofibrate; FCHL: familial combined hyperlipidemia; I: Intralipid; no change: no significant change; SI: sitagliptin; OLO: oral lipid overload; PR-apoB-48: apoprotein B48 production rate; VI: vildagliptin.
Enterocyte involvement in lipid metabolism and intestinal TRL production, recognized as a new component of IR and as a significant element in the etiopathogenesis of atherosclerosis, has justified the search for factors that modulate this production.
Intestinal lipid transportersPassive diffusion was considered for a long time to be the main entry route of nutrients into the intestinal cell. This has changed with the demonstration of specific protein transporters on the microvillus membrane. The main protein transporters include fatty acid translocase (FAT/CD36), involved in the absorption of cholesterol and long-chain fatty acids; Niemann-Pick C1 like 1 (NPC1L1), involved in cholesterol and phytosterol absorption; ATP-binding-cassette transporter A1 (ABCA1), involved in cholesterol flow; and ATP-binding-cassette transporter G5/G8 (ABCG5/G8), involved in sterol flow. Enterocyte lipid content is a regulator of chylomicron production. The rat model deficient in the FAT/CD36 transporter, showing decreased lymphatic secretion of TG, illustrates this condition.65 In rat models of diabetes (induced by streptozotocin), as compared to control rats, intestinal mRNAs of NPC1L1 and MTP increased, while those of ABCG5 and ABCG8 decreased, with a positive correlation with TG and cholesterol levels inside CMs and with apoB-48 levels.4
In a study conducted on type 2 diabetic patients and controls, duodenal biopsy was performed, and mRNAs of NPC1L1, MTP, and ABCG5/8 positively correlated to TG levels, showing an association between the expression of enterocyte levels and lipid abnormalities in type 2 diabetes.66
Free fatty acidsCirculating FFAs are increased in type 2 diabetes, obesity, and FCHL.63 These FFAs accumulate in non-adipocyte tissues, exerting different harmful effects collectively called lipocytotoxicity. FFA flow in the liver stimulates TRL production and secretion,67 mainly in IR, because hepatic oxidation of fatty acids is reduced. Acute elevation of plasma FFA levels stimulates the production of intestinal and hepatic TRLs with no change in clearance in healthy subjects68 (Table 1). This stimulating effect of FFAs on TRL production had previously been shown in the insulin-sensitive Syrian golden hamster model with no additive effect in the insulin-resistant hamster model, except when previously treated with insulin sensitizers such as rosiglitazone.51 The mechanisms accounting for this phenomenon are the blockade of the insulin signaling pathway, the stabilization of apoB-48, the direct incorporation of FFAs into lipoproteins, the stimulation of the mobilization and incorporation of reserve TG, and increased de novo TG synthesis51 (Fig. 2).
An in vivo, ex vivo and in vitro study in rats showed that the effect of FFAs in the liver is bimodal; FFAs at small concentrations and for a short time stimulate the secretion of hepatic TRLs, while FFAs at high concentrations and for a long time decrease the production of hepatic TRLs.69 This suppressive effect of FFAs on hepatic TRL production was confirmed in patients with type 2 diabetes under hyperinsulinemic-euglycemic clamp after the infusion of Intralipid and heparin.40 This bimodal effect of FFAs was absent for apoB-48 in the animal model and in the population with type 2 diabetes.40,69
InsulinIn contrast to the stimulating effect of hyperinsulinism associated with IR on the production of hepatic and intestinal TRLs,4,23 it is commonly accepted that acute elevation of insulin levels inhibits the secretion of hepatic and intestinal TRLs. This has been shown in humans both in vivo70–72 and in vitro.73 In healthy humans, Malmström et al. showed that the inhibitory effect of insulin on hepatic TRL production was independent of the suppressive effect of insulin on FFAs70; by contrast, Pavlic et al. showed that the inhibitory effect of insulin on intestinal TRL production was partly dependent on the effect of insulin on FFAs72 (Table 1).
In healthy humans, hyperinsulinism induced by foods with a high glycemic index or by a hyperinsulinemic-euglycemic clamp delays the appearance of intestinal TRLs in plasma after a fat-rich meal74 (Table 1). An in vitro study showed that the acute addition of insulin to a culture medium of human fetal intestinal cells reduced chylomicron secretion.75 Patients with type 2 diabetes, obese and with FCHL who have chronic hyperinsulinism secondary to IR do not respond to the acute inhibitory effect of insulin on the production of hepatic TRLs, probably secondary to the presence of liver steatosis.71,76,77 Similar checks have been performed in hepatocytes from rats with IR.78 The suppressive action of insulin on CM production is absent in obese patients with IR subject to a hyperinsulinemic clamp.58 We recently showed, in a previous study, that patients with type 2 diabetes under a hyperinsulinemic-euglycemic clamp are refractory to the acute inhibitory effect of insulin on intestinal TRL production40 (Table 2).
GlucagonThis pancreatic hormone may be involved in oxidative FFA metabolism in the liver.79 In rats, glucagon decreased TG synthesis and secretion, and activated β-oxidation through the activation of PPAR-α.79 In healthy humans, acute glucagon administration decreased secretion and catabolism of VLDL type 1, with no changes in their plasma levels; by contrast, no significant changes were seen in CM secretion and catabolism80 (Table 1). The lack of effect of glucagon on enterocytes may be due to the decrease in glucagon receptors in the bowel as compared to the liver.81
IncretinsGlucagon-like peptide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2) are secreted in response to the oral intake of food, particularly food rich in fat and carbohydrates. In humans, GLP-1 secretion may be stimulated by oleic acid,82 which suggests an association between lipid metabolism and GLP-1. The concept that incretins modulate the metabolism of chylomicrons emerged in 2005, when the administration of recombinant GLP-1 to healthy rats was shown to decrease intestinal TG secretion.83 The administration of a dipeptidyl peptidase IV (DPP-IV) inhibitor, such as sitagliptin, decreases TG secretion in the Syrian golden hamster model. This model has made it possible to explain the link between insulin signaling and the effect of GLP-1.84
The administration of GLP-1 to healthy subjects decreased postprandial lipidemia (absence of TG elevation and decrease in plasma FFAs).85 A recent study showed that treatment of healthy men with exenatide (a GLP-1 analog) for 4 weeks decreased PR-apoB-48 without changing PR-apoB-100, suggesting that the bowel is the site of action of GLP-1 in lipid metabolism86 (Table 1).
The use of vildagliptin (a DPP-IV inhibitor) for 4 weeks improved postprandial lipidemia (decreasing plasma TG, chylomicron TG, and intestinal TRL levels) in patients with type 2 diabetes87 (Table 2). Treatment with sitagliptin for 6 weeks decreased postprandial apoB-48 levels with a concomitant improvement in insulin sensitivity in patients with type 2 diabetes88 (Table 2). In addition, the use of exenatide for 2 weeks in patients with type 2 diabetes decreased postprandial TG secretion.89
By contrast, GLP-2 secreted together with GLP-1 exacerbates postprandial lipidemia. GLP-2 is elevated in the streptozotocin-induced rat model of diabetes, and could be implicated in the intestinal hyperplasia associated with diabetes.90 Beyond its intestinotrophic effects, the acute administration of GLP-2 to humans stimulates postprandial lipidemia, probably increasing the intestinal absorption of lipids.91 Recent data in hamsters and mice support the role of GLP-2 in postprandial hyperlipidemia (increased assembly and secretion of intestinal TRLs) due to increased lipid absorption through expression of the glycosylated form of CD36/fatty acid translocase at the enterocyte apical membrane92 (Fig. 2).
InflammationIR is associated with a chronic inflammatory state. However, few studies have addressed the link between inflammation and IR at enterocyte level. In the Syrian golden hamster model, inflammation induced by the infusion of tumor necrosis factor alpha (TNF-α) was shown to induce intestinal IR by decreasing the insulin signaling cascade (Fig. 2). On the one hand, TNF-α decreases the phosphorylation of subunit α of insulin receptor, IRS-1, and Akt, and, on the other hand, increases the MAPK/ERK-1/2 pathway in the postprandial period. This intestinal IR is associated with the overproduction of intestinal TRLs in fasting and postprandial conditions associated with the increased expression of MTP and transporters such as CD36, without modifying the transcription factors involved in de novo lipogenesis, such as SREBP-1c.93
Intervention studiesSeveral nutritional or pharmacological intervention studies have been conducted with the aim of reducing postprandial lipidemia. This review will only focus on clinical trials evaluating the effect of treatment on intestinal TRL production. These studies usually had small sample sizes and modest statistical power.
Effect of circulating free fatty acidsIn an animal model of metabolic syndrome (the gerbil Psammomys obesus), a diet rich in n-3 fatty acids (omega-3) decreased the synthesis of apo-B-48 and the assembly and secretion of intestinal TRLs.94 The addition of treatment with omega-3 fatty acids (3–4g/day) to a high-calorie diet and treatment with a statin (fluvastatin 80mg/day) significantly decreased fasting levels of apoB-48 in a group of 8 with type 2 diabetes with mixed hyperlipidemia.95
Effect of statins, fibrates, and ezetimibeStatin treatment (atorvastatin 20mg/day for 6 weeks) decreased intestinal TRL production in a group of 6 patients with type 2 diabetes and hypertriglyceridemia. A similar but not significant trend was seen with fibrate treatment (fenofibrate 200mg/day for 6 weeks). However, treatment with fenofibrate increased catabolism of TRL-apoB-48, which does not occur with atorvastatin96 (Table 2). Treatment with atorvastatin (10–80mg/day for 16 weeks) decreased TRL-apoB-48 levels by stimulating clearance in the postprandial state in patients with FCHL97 (Table 2). The use of fenofibrate (200mg/day for 6 months) decreased levels of intestinal TRL remnants in patients with FCHL.98
In a group of 8 patients with hypercholesterolemia, treatment with ezetimibe for 8 weeks did not change the metabolism of intestinal TRLs.99 However, when simvastatin (40mg/day for 6 weeks) was combined with ezetimibe in a group of 16 patients with mixed hyperlipidemia, a significant reduction occurred in the production of intestinal TRLs. This reduction is not achieved with simvastatin or ezetimibe alone.100
Evaluation of triglyceride-rich lipoproteins of intestinal origin in clinical practiceThe intestinal overproduction of TRLs represents a new component of IR which contributes to increased plasma TG levels in both the postprandial and postabsorptive states. Enterocytes are resistant to the suppressive action of insulin on CM production in type 2 diabetes and obesity.40,56 The assessment of postprandial lipidemia in IR states ought to be an additional component of routine examinations, in an attempt to detect more marked subclinical hyperlipidemia in the postprandial state as compared to under fasting conditions. The standardization of the amount of fat needed to stimulate CM metabolism continues to be controversial.42 A recent meta-analysis of 113 clinical trials of postprandial lipidemia proposed the use of a new index, the so-called lipemic index (LI), for which 50–70g of fat per meal are given and postprandial lipidemia is assessed through the AUC 4h after the end of OLO.101,102
Testing of fasting and postprandial apoB-48 levels should be a part of annual examinations in IR states such as type 2 diabetes, obesity, FCHL, and coronary disease due to their association with atherosclerosis.59,103 ApoB-48 would also be helpful for follow-up in treatments with statins, fibrates, and omega-3.95,97 The value of apoB-48 in the clinical and research areas is clearly evident.
ConclusionsThere are many arguments suggesting that the bowel is not a passive organ, but rather a metabolically active organ that receives information from the periphery and is able to modulate its own lipid synthesis and secretion processes in relation to substrates, hormones, and other endogenous or exogenous substances. Functional kinships exist between the bowel and liver. The direct and/or indirect role(s) of enterocyte IR, as well as the cellular and molecular mechanisms of action of these different regulatory factors, have not been elucidated yet. Testing of apoB-48 as a marker of postprandial lipidemia and atheromatosis should be used in the clinical practice of endocrinology.
The atherogenic potential of chylomicrons makes understanding their overproduction in the bowel and their accumulation in plasma in type 2 diabetes, and also in IR syndromes mandatory.
Conflicts of interestThe authors state that they have no conflicts of interest.
Authors thank Dr. León Litwak for his reading and corrections on this study.
Please cite this article as: Nogueira JP, Brites FD. Rol del enterocito en la dislipemia de la resistencia insulínica. Endocrinol Nutr. 2013;60:179–89.
