




Bacille Calmette-Guérin (BCG) vaccination is included in the immunisation schedule for tuberculosis endemic countries with a global coverage at birth close to 90% worldwide. BCG was attenuated from Mycobacterium bovis almost a century ago and provides a strong protection against disseminated forms of the disease, though very limited against pulmonary forms of tuberculosis, responsible for transmission. Novel prophylactic tuberculosis vaccines are in clinical development either to replace BCG or to improve its protection against respiratory forms of the disease. There are limitations understanding the immunological responses involved and the precise type of long-lived immunity that new vaccines need to induce. MTBVAC is the first and only tuberculosis vaccine candidate based on live-attenuated Mycobacterium tuberculosis in clinical evaluation. MTBVAC clinical development plans to target tuberculosis prevention in newborns, as a BCG replacement strategy, and as secondary objective to be tested in adolescents and adults previous vaccinated with BCG.
La vacunación con BCG (bacilo Calmette-Guérin) está incluida en el calendario de inmunización al nacimiento en países con alta incidencia de tuberculosis, con una cobertura global cercana al 90%. BCG tiene casi cien años de antigüedad y está basada en una cepa atenuada de Mycobacterium bovis, proporcionando protección contra las formas diseminadas de la enfermedad pero confiriendo una protección muy limitada contra las formas pulmonares de tuberculosis, responsables de su transmisión. Diferentes vacunas profilácticas contra la tuberculosis se encuentran hoy en desarrollo clínico para reemplazar a BCG o para mejorar la protección en individuos ya vacunados con BCG. MTBVAC es la primera y única vacuna candidata basada en una cepa de Mycobacterium tuberculosis atenuada en evaluación clínica. Los planes de desarrollo clínico del MTBVAC se dirigen en primer lugar a la prevención de la tuberculosis en recién nacidos, para reemplazar a BCG, y en segundo lugar en adolescentes y adultos.
Tuberculosis is the infectious disease that accounts for the most deaths worldwide—even more than AIDS. According to the latest estimates from the World Health Organisation (WHO), for 2016, 10.4 million new cases of tuberculosis caused around 1.7 million deaths.1 Tuberculosis is linked to poverty and worsened by the HIV/AIDS pandemic. At present, the emergence of multidrug-resistant strains is one of the greatest threats. Fifty million people are infected with multidrug-resistant strains of Mycobacterium tuberculosis, creating a reservoir for future cases of active tuberculosis. This represents a tremendous barrier to treatment.1 Effective management of tuberculosis requires discovery of faster, more reliable diagnostic tools than those available now; new drugs that shorten treatment duration; and new, more effective vaccines than the current BCG vaccine against the pulmonary forms of the disease, which are responsible for its transmission.1
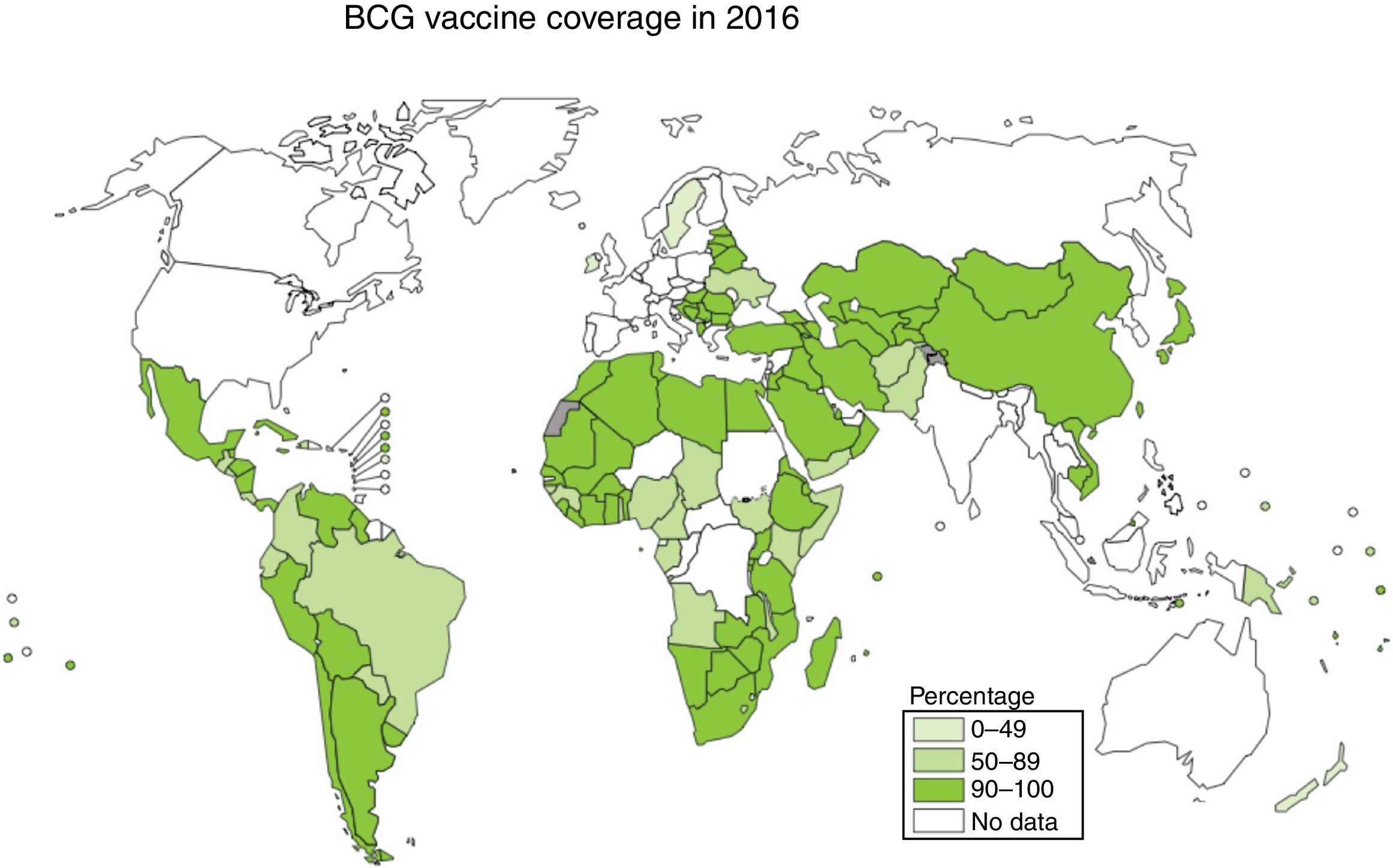
Vaccination is one of the most effective measures to combat infectious diseases in terms of cost–benefit ratio. Bacille Calmette-Guerin (BCG) is currently the only vaccine authorised and in use against tuberculosis, with coverage approaching 90% in countries with a high incidence; however, its efficacy against the respiratory forms of the disease is highly variable2,3 (Fig. 1).
History of BCG
BCG is a live attenuated vaccine derived from Mycobacterium bovis, the causative agent of tuberculosis in cattle.4 BCG was developed by Albert Calmette, a physician, and Camille Guérin, a veterinary surgeon, between 1908 and 1921, by repeated subculture of a strain of M. bovis isolated from a cow. Calmette and Guérin followed Pasteur's principles for the construction of live attenuated vaccines against infectious diseases. Following 230 passes in the laboratory over the course of 13 years, the strain demonstrated attenuation first in calves and then in guinea pigs and other animal models.5 BCG was first introduced in a clinical setting nearly a hundred years ago, in 1921, when it was administered orally to a baby whose mother had died of tuberculosis the day after the baby was born. The baby did not have any adverse effects due to BCG vaccination and, most importantly, did not develop tuberculosis. At that time, the oral route of administration was selected for BCG, as the gastrointestinal tract was believed to be a natural route by which infants and children given unpasteurised milk were infected with tuberculosis.6 Between 1921 and 1926, more than 50000 children were vaccinated and experienced hardly any adverse effects. The mortality rate among vaccinated children was 1.8%, versus the mortality rate among unvaccinated children of over 25%. Thus, the vaccine demonstrated its efficacy as it decreased child mortality due not only to tuberculosis.5
The original BCG strain had spread throughout the world before being preserved by means of lyophilisation in the 1960s. Given that there were no methods for long-term preservation of microorganisms up to that time, individual laboratories performed repeated passes in which they subcultured the original strain. This led to the emergence of different BCG substrains which were named by the name of the laboratory or country where they were subcultured, resulting in different BCGs with heterogeneous phenotypes. At present, six strains are most often used in international immunisation programmes worldwide: BCG Pasteur 1173 P2, BCG Danish 1331, BCG Glaxo 107, BCG Tokyo 172-1, BCG Russia-Ir and BCG Brazil.7
The main cause of BCG attenuation is loss of region of difference 1 (RD1) associated with loss of the 6-kDa immunodominant secreted antigen (ESAT-6) virulence factor.4 Genomic analysis has shown multiple differences between BCG substrains, including deletions other than deletion of RD1 which contribute to phenotypic variations among them. There are clear differences in attenuation, but these have not been shown to contribute to differences in efficacy.8
Vaccination with BCG todaySince 1974, intradermal BCG vaccination at birth has been included in the WHO Expanded Programme on Immunisation (EPI). This has resulted in the administration of more than 4 billion vaccines worldwide, with approximately 200 million vaccines being administered every year. Levels of live bacteria in vaccines range from 50000 to 3 million per dose, depending on the BCG strain used.9
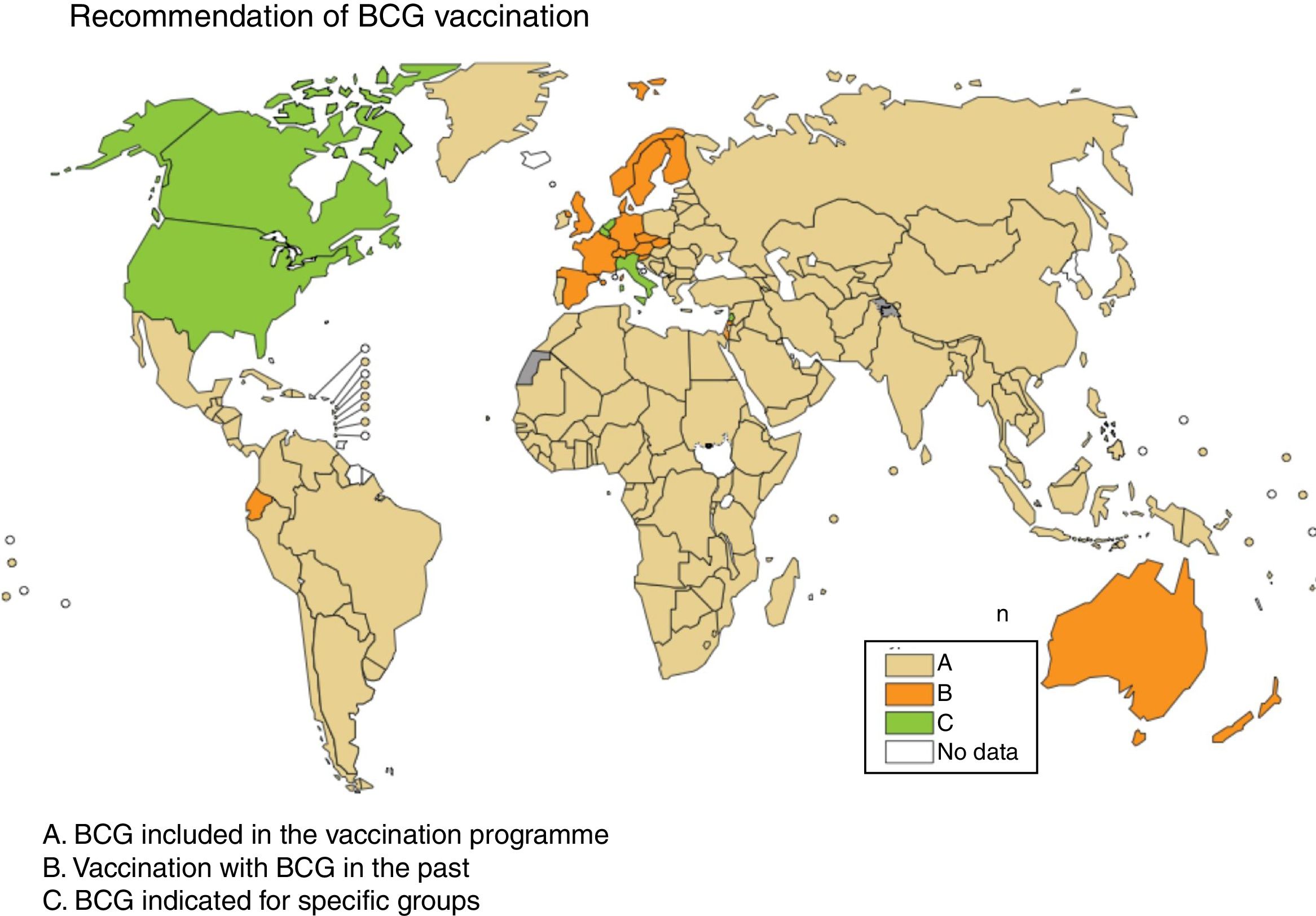
Some European countries, such as Ireland, currently recommend BCG vaccination, whereas other countries, such as France and Portugal, used to recommended BCG vaccination but no longer do (Fig. 2). Information on BCG vaccination policies and practices worldwide may be viewed at http://www.bcgatlas.org/. Specific information for Europe may be viewed at http://www.vaccine-schedule.ecdc.europa.eu/Pages/Scheduler.aspx.
, the countries where BCG vaccination used to be but is no longer done are in orange (B) and the countries where BCG is indicated only in specific population groups are in green (C). Source: WHO Global Tuberculosis Report 2017.")
Recommendation of BCG vaccination. The countries where BCG is included in the vaccination programme are in beige (A), the countries where BCG vaccination used to be but is no longer done are in orange (B) and the countries where BCG is indicated only in specific population groups are in green (C). Source: WHO Global Tuberculosis Report 2017.
Today, the WHO recommends BCG vaccination in all newborns in countries with a high incidence of tuberculosis. BCG is not recommended for babies with known HIV infection or any other immunodeficiency. In countries with a high prevalence of tuberculosis and HIV, it is important to be careful when routinely administering BCG due to a risk of disseminated BCG disease in HIV-infected babies. BCG is recommended in asymptomatic infants born to mothers of unknown HIV status. Today, it is recommended that routine BCG immunisation during childhood be done until a systematic HIV screening programme can be implemented.
In countries with a low incidence of tuberculosis, BCG immunisation can be considered in children ≤5 years of age from endemic countries. It can also be considered in a child continuously exposed to a patient with infectious pulmonary tuberculosis who has not been treated or not responded to treatment when the child cannot be separated from the patient. Finally, it can be considered in a child continuously exposed to a patient with infectious pulmonary tuberculosis caused by M. tuberculosis strains resistant to isoniazid and rifampicin when it is impossible to separate the child from the infectious patient.10
The International Union Against Tuberculosis and Lung Disease (IUATLD) and the WHO have established a number of criteria under which they recommend that a country switch from a systematic BCG vaccination policy to selective vaccination of high-risk groups. Stopping systematic BCG vaccination in a certain country would be recommended in the event that, if there is an effective system for reporting tuberculosis cases, the annual rate of reporting of pulmonary tuberculosis with bacilloscopy is lower than 5 per 100000 inhabitants, or the annual risk of tuberculosis infection is <0.1%. It would also be recommended if the annual rate of reporting of tuberculosis meningitis is <1 per 10 million inhabitants in the last 5 years.10
The recommendation of BCG vaccination for adults who travel to endemic areas with high exposure to multidrug-resistant tuberculosis remains controversial. Given the potential risk of failure of antituberculosis treatment as well as the low rate of complications related to BCG vaccination in immunocompetent individuals, BCG administration could be recommended in non-vaccinated individuals with a negative tuberculin skin test and negative IGRAs who are exposed to multidrug-resistant tuberculosis. More studies are needed to verify the protective efficacy of BCG in the context of exposure to multidrug-resistant tuberculosis in adults.10
The safety of the BCG vaccine has been widely proved as more than 4 billion units have been administered worldwide since 1921. BCG is a highly reactogenic but very safe and well-tolerated vaccine that causes a local reaction at the injection site.9 Some 2–6 weeks after the vaccine is administered, a small papule appears that increases in size and in most cases progresses to an ulcer. The cervical and axillary lymph nodes may temporarily enlarge. After approximately 3 months, a permanent scar will develop.
Contraindications for BCGBCG at birth would be contraindicated in malnourished children and premature newborns with a birth weight of less than 2.5kg. Given that the BCG vaccine is a live vaccine, it is also contraindicated in pregnant women as well as oncology patients and immunocompromised patients who suffer from congenital immunodeficiency or immunodeficiency acquired though immunosuppressants, radiation or HIV infection. In addition, it is not recommended that BCG vaccine be administered if a patient has been treated with antibiotics in the last 30 days.
BCG is also contraindicated in people with active tuberculosis or a positive tuberculin skin test, or with tuberculosis infection; however, recent studies have shown that BCG is safe in people with latent tuberculosis infection.11
Administration with other vaccines and productsBCG improves the responses of T and B cells to other vaccine antigens and may be co-administered with any other vaccine (including other live vaccines) with no major problems having been reported. BCG improves the responses of Th1 and Th2 cytokines to unrelated antigens and increases the response of antibodies in oral vaccination against polio.
The only limitations would be the need for administration in different anatomical sites and the fact that it is not recommended that any other vaccine be administered in the same limb in the 3 months following administration of BCG.10
Immune response to BCGWhile cellular immunity is known to be important for managing tuberculosis, no immunological marker can be correlated with protection against the disease. Immune response to primary BCG immunisation has been evaluated in different studies in children. These studies have shown a BCG-associated induction of polyfunctional CD4+ and CD8+ T cells, interferon (IFN-γ)+, interleukin (IL)-2+ and tumour necrosis factor (TNF-α)+. However, it has not been possible to demonstrate a correlation in terms of protection.12
Variable efficacy of BCGDespite its broad coverage, the extent to which BCG protects against the respiratory forms of tuberculosis remains subject to debate.3 In infants, BCG has been shown to offer protection against disseminated forms of tuberculosis (miliary and meningeal).13 In adolescents and adults vaccinated at birth, the efficacy of BCG against the pulmonary forms of the disease is highly variable, depending on the age at which individuals are vaccinated and assessed.2 BCG immunity is believed to decrease over time. It is recommended that the vaccine be administered as close to birth as possible.14
Recent studies have shown that BCG, despite conferring moderate protection, lasts longer than previously believed—up to at least 20 years.15 Hypotheses as to why BCG efficacy is so inconsistent are many and varied. None of them has led to a definitive answer. It is believed, but not proved, that BCG overattenuation due to a loss of immunodominant antigens during the process of repeated subcultures could be one of the reasons for these differences, as mentioned above.8 Many clinical trials have suggested that BCG revaccination does not improve BCG efficacy. This could be due to pre-existing immunity arising from infection with non-tuberculous environmental mycobacteria prior to vaccination which could result in masking or blocking of BCG revaccination.16
Non-specific benefits of BCG vaccinationGrowing numbers of studies have shown that live attenuated vaccines reduce child morbidity and mortality. Today, this is a fact admitted by the WHO.17,18 In some countries in western Africa, BCG reduces neonatal mortality by more than 40%, mainly through prevention of sepsis, respiratory infections and fever.19 Studies in countries where child mortality is very low, such as Spain, have found BCG vaccination at birth to have non-specific benefits, concluding that it decreases hospitalisation due to respiratory infections and sepsis not related to tuberculosis through a non-specific effect.20,21 It has recently been shown that BCG may induce non-specific resistance to pathogens through epigenetic reprogramming of monocytes.22,23 These effects have been reported for innate immune system cells, such as macrophages and natural killer (NK) cells.24,25 Metabolic pathways play an essential role in immunity in human monocytes, regulated by epigenetic mechanisms on a level of chromatin organisation, and highlight the therapeutic potential of modulation of these pathways during vaccination.26 Any new vaccine with better efficacy against the respiratory forms of tuberculosis must also feature these non-specific effects exhibited by BCG.
Research and development of new vaccines against tuberculosisGiven BCG's lack of protection against the respiratory forms of tuberculosis, tremendous efforts have been invested in research and development for new vaccines against tuberculosis in the last 20 years.27 After thousands of candidates were discovered and hundreds proceeded to preclinical trials in animal models, less than a hundred went on to be trialled in clinical studies in humans. Each candidate vaccine must go through different steps in phase I, phase II and phase III clinical trials to obtain a marketing authorisation. In Europe, research driven by the different Framework Programmes of the European Commission has enabled hundreds of candidates to proceed to preclinical trials. Several are currently in clinical trials in humans.28
The most effective vaccines in use today against different infectious diseases induce neutralising antibodies, thereby conferring protective immunity. For other diseases such as AIDS, malaria and tuberculosis, a strong cellular immune response is needed.29
One of the greatest difficulties involved in finding effective vaccines against these diseases is that no immunological marker can predict the efficacy of a new vaccine being studied. This means that new vaccines must be tested in long and costly efficacy studies with thousands of volunteers (phase IIb and phase III) in endemic countries with high incidences of these diseases after robust safety and immunogenicity data have been obtained in prior trials with tens (phase I) and later with hundreds (phase II) of healthy volunteers.27
First clinical trial of efficacy of a new vaccine against tuberculosisFollowing more than 10 years of prior clinical trials, the MVA85A vaccine, developed by the University of Oxford and led by Dr Helen McShane, was the subject of the first clinical trial of efficacy of a vaccine against tuberculosis in an endemic country, in Worcester, South Africa.30 The MVA85A vaccine was developed to increase immunity in children previously vaccinated with BCG. These children were administered modified vaccinia virus Ankara (MVA) into which the gene that codes for the major tuberculosis antigen Ag85A had been inserted. The phase IIb efficacy study consisted of a double-blind, placebo-controlled study in healthy non-HIV infected children 4–6 months of age who had received BCG at birth, with a follow-up every 3 months for more than 3 years. A total of 2797 children were vaccinated (1399 with MVA85A and 1398 with a placebo). The results showed that 32 children (2%) out of the 1399 children vaccinated with BCG+ MVA85A were diagnosed with tuberculosis, whereas 39 children (3%) out of the 1398 children vaccinated with BCG+ placebo were diagnosed with the disease. The difference between the two groups was not significant. The results of the study were interpreted to mean that the MVA85 vaccine lacked efficacy.30 They were considered a failure by the agencies that funded the study.
Yet, for the scientific community working on vaccines against tuberculosis, they represented a major step forward in research on new vaccines, since this trial opened the door to further efficacy studies. The MVA85 efficacy trial was the first in nearly a hundred years after BCG in the 1920s. The Worcester study was coordinated by the South African Tuberculosis Vaccine Initiative (SATVI). Years after this study, the scientific community continues to learn and draw conclusions from it on the immunology of the disease. After the children in the study had been followed up for 3 years, QuantiFERON (QFT) test conversion and disease risk were studied. These results30 showed that in children with a negative QFT test (<0.35IU/ml) and in children with a positive QFT test but with less than 4IU/ml, the risk of developing tuberculosis was low, whereas in children with a positive QFT test with >4IU/ml, the risk was high.31
Diversity of new candidate vaccines in clinical trialsFollowing the publication of the results of the first efficacy study in children, funding agencies and researchers raised the question of how to diversify candidate vaccines against tuberculosis, since most current candidates were vaccines with little diversity of antigens (Ag85A or Ag85B, ESAT6) designed to enhance prior T cell-mediated immunity.27 In the last 10 years, two organisations have assumed responsibility for coordinating the search for new candidate vaccines in different preclinical phases and accelerating the use in humans of new vaccines developed in laboratories. One was the TuBerculosis Vaccine Initiative (TBVI), a European organisation. The other was the Aeras Global TB Vaccine Foundation, an American organisation backed by the Bill and Melinda Gates Foundation.
Which population should we vaccinate?After the first efficacy study yielded negative results, a question had to be answered. Which population took priority with respect to vaccination: children or adults? Modelling studies have shown that the greatest impact of a new vaccine against tuberculosis would clearly be in adolescents and adults, where the transmission of the disease is greater32, since, although the incidence of the disease is very significant in children under 5 years of age, the respiratory forms responsible for transmission are not common in them.33
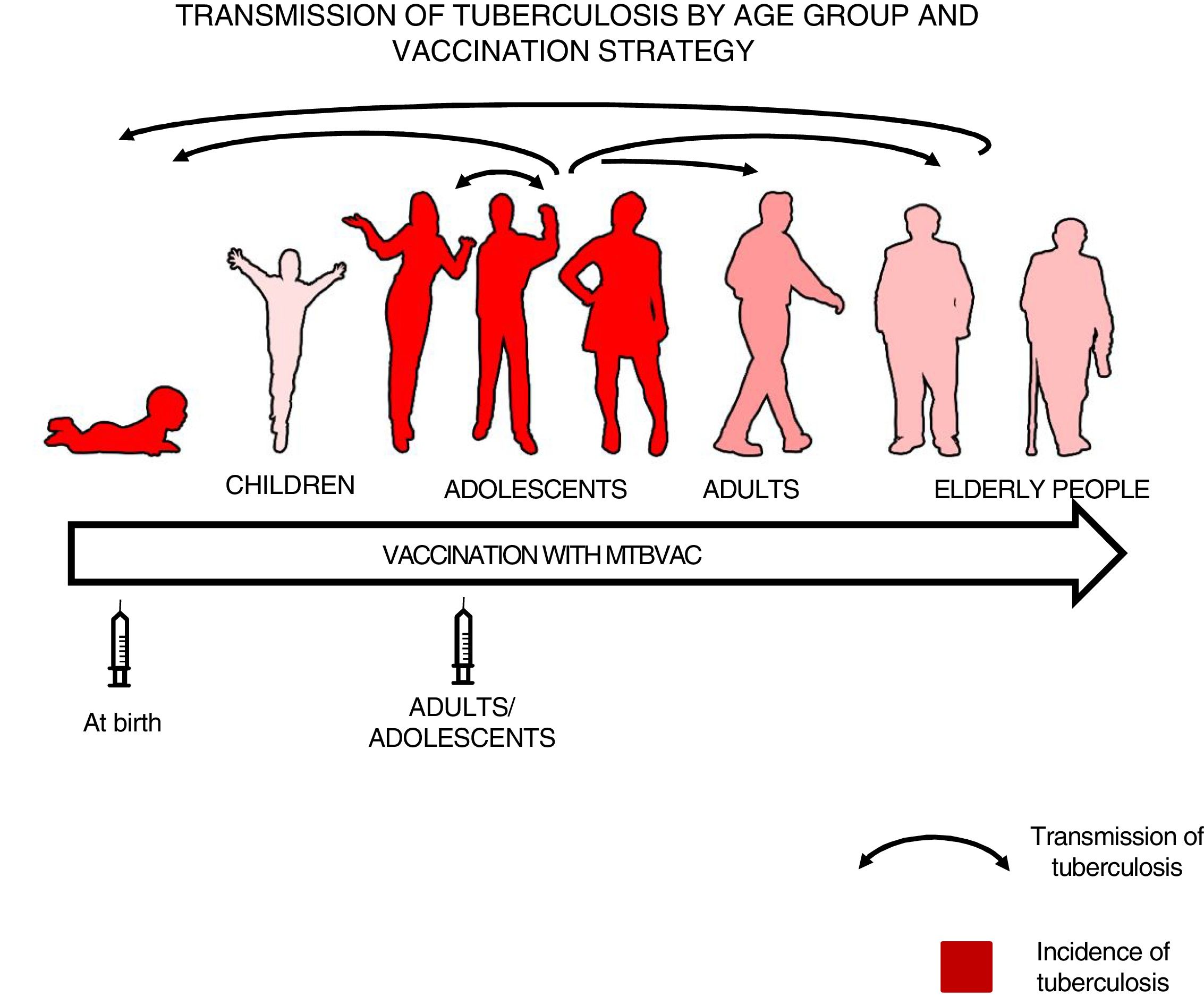
We felt that healthy newborns represent the most sensitive population to trial the efficacy of a new vaccine as they have no pre-existing immunity to BCG or environmental mycobacteria, whereas trialling a new vaccine in an older population could result in masking or blocking of vaccination.16,34 Therefore, we thought it best to first study efficacy in newborns to attempt to demonstrate the efficacy of a new vaccine, then vaccinate adolescents and adults to have a greater impact on tuberculosis as pulmonary forms are responsible for the transmission of the disease34 (Fig. 3).

Transmission of tuberculosis by age group and vaccination strategy. The role of adolescents and adults in the transmission of the disease is indicated by arrows. The groups with the highest incidence of tuberculosis—children under 5 years of age and adolescents—are in red. The introduction of a new vaccine at birth would enable the protection of children from birth and the study of its efficacy in a naïve population neither previously exposed to mycobacteria nor previously vaccinated with BCG. Vaccination in an adolescent and adult population with pulmonary forms of tuberculosis would have a greater impact on the transmission of the disease.
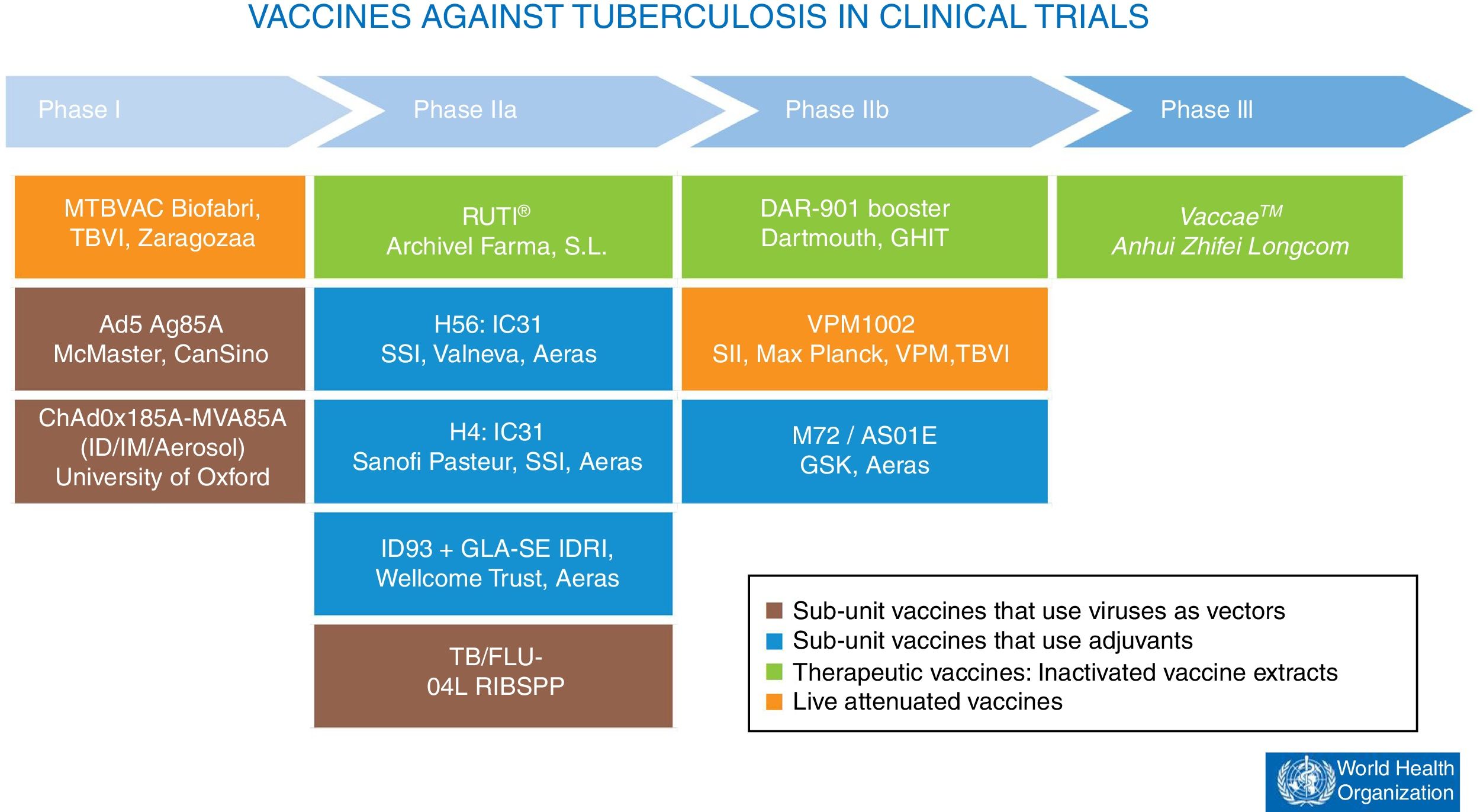
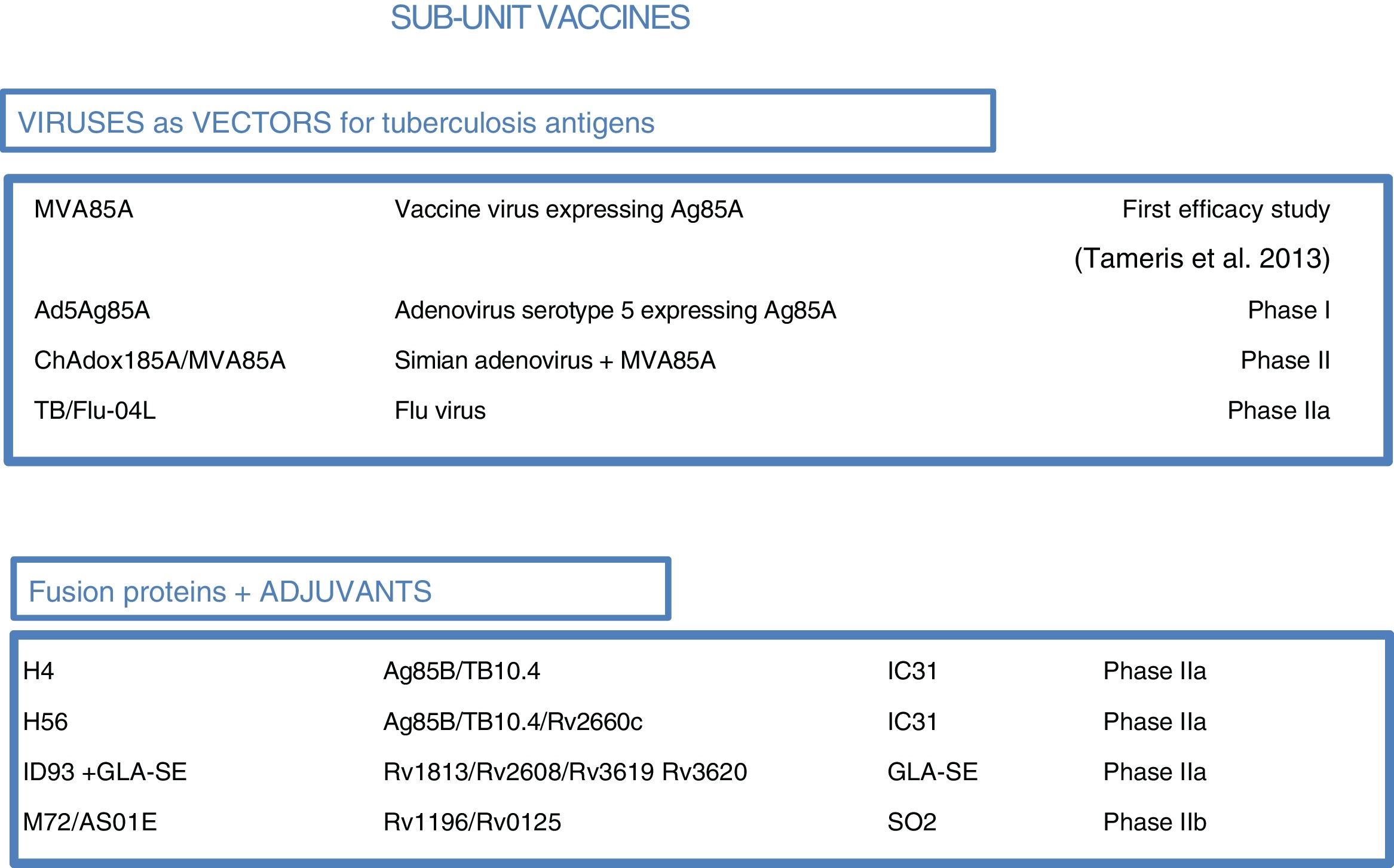
At present, there are 12 vaccines against tuberculosis in the clinical trial phase1 (Fig. 4). Most of these vaccines are based on sub-units in which M. tuberculosis antigens are expressed as recombinant proteins formulated with different adjuvants or expressed through recombinant viruses used as vehicles for administering antigens.

Vaccines against tuberculosis in clinical trials. Sub-unit vaccines that use viruses as vectors are in brown. Sub-unit vaccines that use adjuvants for their administration are in blue. Therapeutic vaccines that consist of inactivated vaccine extracts are in green. Live attenuated vaccines are in orange. Source: WHO Global Tuberculosis Report 2017.
In clinical practice today, some of these vaccines are based on inactivated mycobacteria and were designed as “therapeutic vaccines” with the hope of decreasing treatment times in people infected with latent tuberculosis or reducing the likelihood of recurrence after the end of treatment.35 The two therapeutic vaccines in clinical development consist of either inactivated forms of non-tuberculous mycobacteria, as in Vaccae™, which is in phase III efficacy studies, or M. tuberculosis cell wall fragments transported in liposomes, as in the RUTI® vaccine, which is currently in phase IIa clinical trials.36
Classic vaccination strategies seek to prevent disease through so-called prophylactic vaccines. In tuberculosis, two strategies have been developed to develop preventive vaccines. The first aims to enhance the action of the current BCG vaccine by boosting the protection it confers. The second aims to replace the current BCG vaccine with a more effective one.
As most of the population of countries where tuberculosis is endemic was vaccinated with BCG at birth, booster vaccines with M. tuberculosis-specific antigens seek to enhance BCG. One such vaccine in a clinical trial, DAR-901, is based on a non-tuberculous bacterium inactivated by heat.1,37 Other sub-unit vaccines include just one or a few M. tuberculosis-specific protein antigens and are administered in viral vectors or formulated with adjuvants1 (Fig. 5).
, adenoviruses of different origins (Ad or ChA) and the flu virus. Other sub-unit vaccines use different adjuvants (IC31, GLA-SE or SO2) to enhance the immunogenic effects of M. tuberculosis proteins. The current phase of clinical development is indicated for each candidate sub-unit.")
Sub-unit vaccines in clinical trials. Sub-unit vaccines seek in individuals previously vaccinated with BCG to increase the protection it confers by boosting it with M. tuberculosis antigens. They may use different viruses as vectors, such as poxviruses (MVA), adenoviruses of different origins (Ad or ChA) and the flu virus. Other sub-unit vaccines use different adjuvants (IC31, GLA-SE or SO2) to enhance the immunogenic effects of M. tuberculosis proteins. The current phase of clinical development is indicated for each candidate sub-unit.
Three sub-unit vaccines use viral vectors with different viruses and different routes of administration. One, currently in phase IIa, is the TB/FLU-04L vaccine. This uses the attenuated flu virus as a recombinant vector and expresses Ag85A and ESAT6 antigens. It is administered through the mucosa. Two others, the Ad5 Ag85 and ChadAdOx18A-MVA85A vaccines, are currently in phase I clinical trials. The Ad5 Ag85A vaccine is being developed by McMaster University in Canada. It consists of a serotype 5 adenovirus vector that expresses Ag85A and is administered intramuscularly. The ChAdOx185A vaccine, developed by the University of Oxford, is based on a simian adenovirus and MVA85A (modified smallpox virus). It expresses Ag85A systemically and through the mucosa.
Four other sub-unit vaccines use different adjuvants to administer tuberculosis antigens. M72/AS01E, developed by Glaxo, is in phase IIb clinical trials. Three others are in phase IIa clinical trials: the H4:IC31 and H56:IC31 vaccines, developed by the Statens Serum Institut (SSI) in Copenhagen, and the ID93+ GLA-SE vaccine, developed by the Infectious Disease Research Institute (IDRI) in the United States.
M72/AS01E is a sub-unit vaccine that combines two M. tuberculosis antigens (32A and 39A) and an adjuvant (AS01E). It is being tested in a phase IIb trial of efficacy in HIV-negative adults infected with M. tuberculosis in Kenya, South Africa and Zambia. H4:IC31 is a BCG booster vaccine that contains an Ag85B–TB10.4 fusion protein formulated with adjuvant IC31.
The IC31 H56:IC31 vaccine combines three M. tuberculosis antigens (Ag85B, ESAT-6 and Rv2660c) and adjuvant IC31. The ID93+ GLA-SE vaccine contains four M. tuberculosis antigens associated with virulence (Rv2608, Rv3619 and Rv3620) and latency (Rv1813) as well as adjuvant GLA-SE.
In prophylactic vaccines that seek to replace BCG, the immunity conferred by live vaccines is believed to induce long-lasting specific memory immune responses not achieved with sub-unit vaccines. This effect could be related to the persistence or limited replication in vivo seen for other live human vaccines (e.g. polio, measles and yellow fever).38
Two vaccines seek to replace BCG at birth. They are based on live attenuated vaccines. One is the VPM1002 vaccine, derived from M. bovis BCG, developed by the Max Planck Institute in Berlin and in phase IIa clinical trials. The other is MTBVAC, derived from M. tuberculosis, in phase I clinical trials and having started a phase IIa clinical trial in babies and for adults at SATVI, in South Africa, in 2018.
The VPM1002 vaccine is based on recombinant BCG (rBCG), expresses Listeria monocytogenes listeriolysin and features deletion of the gene that codes for urease C. It is designed to improve the efficacy of BCG through insertion of other genes. At present, a phase II clinical trial is under way in South Africa to evaluate the safety and immunogenicity of the vaccine in HIV-exposed and non-HIV-exposed newborns.39
Clinical studies of efficacy of MTBVAC: a live attenuated vaccine from the human pathogenAt present, the immune responses and the precise type of lasting immunity that a new vaccine against tuberculosis should induce are unknown. This means that effective vaccines have to be developed in order for the immunology of protection against the disease to be understood. As mentioned above, most vaccines are mainly based on a small number of antigens with different systems for administration. Therefore, with the hope of increasing the diversity of candidate vaccines, we chose to construct a live attenuated vaccine from a pathogen of human origin in pursuit of imitating natural infection. We did so because close to 80% of those with latent tuberculosis infection do not experience reinfection with M. tuberculosis40 and because the risk of developing tuberculosis is eliminated as the strain is attenuated. In addition, we were able to maintain the complete antigen repertoire of the human pathogen as we started with a live bacterium.
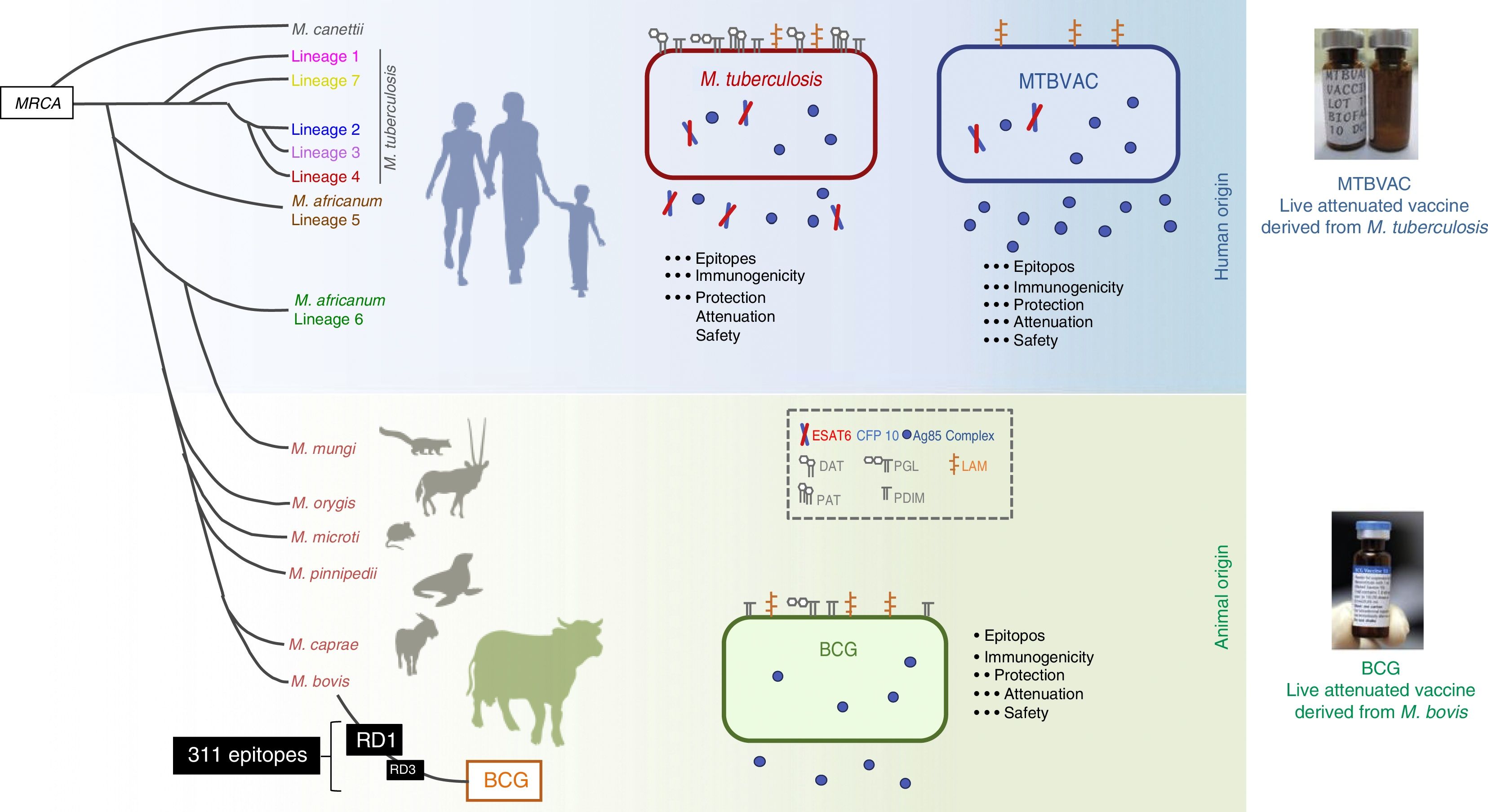
The MTBVAC vaccine is the only live attenuated vaccine derived from an isolate of M. tuberculosis in clinical trials meeting the requirements set out in the Geneva Consensus41,42 (Fig. 6). Developed by the Universidad de Zaragoza and the Institut Pasteur with the support of the TuBerculosis Vaccine Initiative (TBVI), MTBVAC showed a longer-lasting T cell response to different M. tuberculosis antigens not present in BCG43 and conferred better protection than BCG in various animal models.44,45 Industrial development and subsequent clinical development were done by Biofabri, a Spanish biopharmaceutical company with extensive experience in live attenuated vaccines (CZ veterinary surgeon). Subculture was avoided so as not to give rise to different strains, as occurred with BCG in the past.

Differences between MTBVAC and BCG. The figure shows a representation of M. tuberculosis, BCG and MTBVAC. BCG has lost RD1 which has led to its attenuation. MTBVAC features the antigen repertoire of M. tuberculosis absent in BCG including RD1. It contains more than 300 epitopes and at the same time presents an attenuation profile comparable to that of BCG.
MTBVAC was constructed from a human isolate of M. tuberculosis belonging to lineage 4 (Euro–African–American), one of the most widespread lineages of M. tuberculosis. The vaccine consists of two stable mutations through deletion of the phoP and fadD26 virulence genes. Therefore, MTBVAC contains all the genes present in M. tuberculosis strains, including the genes absent in M. bovis and deleted during BCG subcultures. MTBVAC has undergone its first clinical trial in humans.46 The results of the phase Ia clinical trial in adults showed a robust safety profile for MTBVAC in the three doses tested up to a dose of 105, which is equivalent to that currently used with BCG.47 Immunology studies done by stimulating participants’ blood with BCG or MTBVAC showed a dose–response correlation for polyfunctional CD4 lymphocytes. In the volunteers who received the same dose of MTBVAC as of BCG, the number of individuals who responded 4 weeks following vaccination was higher in the group vaccinated with MTBVAC.
The ELISpot test for ESAT6 and CFP10, used to distinguish between individuals vaccinated with BCG and individuals infected with tuberculosis, was negative for everybody vaccinated with MTBVAC47 7 months after vaccination. However, a trend in specific response against CFP10-ESAT 6 could be seen in participants vaccinated with MTBVAC compared to BCG.42
One of the main differences between the BCG and MTBVAC vaccines is that many epitopes absent in BCG are present in MTBVAC. MTBVAC may be said to possess 50% more epitopes recognised by human T cells compared to BCG. In addition, MTBVAC secretes more Ag85 complex proteins than BCG48 and, unlike BCG, secretes Ag85B. MTBVAC produces the main antigens deleted in all BCG strains and present in the RD1 region, such as ESAT6 and CFP10.42 Our recent studies have shown the importance of host recognition of these antigens. Only mice with major histocompatibility complexes (MHCs) capable of recognising ESAT6/CFP10 are better protected against infection. This has demonstrated that the protective efficacy of MTBVAC is associated with T cell-mediated response to CFP10/ESAT-6, which may be important for immunity.49
The clinical development plan for MTBVAC considers vaccination in newborns its primary objective, as this population has not been previously exposed to environmental mycobacteria or individuals previously vaccinated with BCG. It is important to stress the importance of vaccination of this population given the high incidence of tuberculosis in babies under 5 years of age (very similar to adolescents).33
Today, the phase Ib clinical trial in babies is ending at SATVI in Worcester, where newborns were vaccinated with increasing doses of MTBVAC and vaccination with the highest dose ended in September 2016. After more than a year of follow-up, no major undesirable effects related to the vaccine have been reported. Immunology results are expected to be ready in late 2018 (ClinicalTrials.gov Identifier: NCT02729571).
Future clinical studies of efficacy of MTBVACThe European Union, through its European and Developing Countries Clinical Trials Partnership (EDCTP) programme, recently approved funding for a phase IIa clinical trial in babies. This phase IIa clinical trial, intended to find an optimal dose in newborns (non-HIV-exposed, BCG with no prior treatment, no known family exposure to tuberculosis), has the primary objective of evaluating the safety and reactogenicity of MTBVAC in increasing doses compared to the BCG vaccine and to evaluate the immunogenicity of MTBVAC with three increasing doses compared to the BCG vaccine. The secondary objective is to evaluate the dynamics of QFT conversion and reversion induced by the MTBVAC vaccine. A total of 99 newborns will be vaccinated (75+24). In cohort 1, each different dose of MTBVAC will be administered to 25 newborns and BCG will be administered to 24 newborns. Epidemiology will be studied to prepare future efficacy trials at two additional sites: the Institut Pasteur in Madagascar and Saint-Louis, Senegal.
The secondary objective for the clinical development of MTBVAC is to vaccinate adolescents/adults. The study, recently approved by the United States Congress and National Institutes of Health (NIH) and coordinated by Aeras, will commence in 2018. The trial will consist of a safety, immunogenicity and dose-scaling study in adults with or without latent tuberculosis infection, also in South Africa (ClinicalTrials.gov Identifier: NCT02933281). The primary objective is to study the safety and reactogenicity of MTBVAC compared to BCG in adults. The secondary objective is to study the immunogenicity of MTBVAC at four increasing doses measured through complete blood testing and QuantiFERON conversion rates in QFT-negative adults.
Live vaccines such as MTBVAC are solid candidates for “replacing” BCG and for being used where they are needed most—in countries with the highest incidences of tuberculosis—once MTBVAC can be shown to be better than BCG. The challenge at hand consists of planning clinical trials of efficacy in countries with high incidences of tuberculosis. This could be accelerated to a large extent by identifying markers of protection.
FundingThis study was funded by the Spanish Ministry of Economy and Competitiveness (BIO2014-5258P) as well as the European Union as part of its H2020 programme (TBVAC2020 643381) and the European & Developing Countries Clinical Trials Partnership (EDCTP) (RIA2016V-1637).
Conflicts of interestC.M., J.G. and N.A. are inventors of tuberculosis vaccine-related patents. The Universidad de Zaragoza is the holder of these patents, and the Spanish biotechnology company Biofabri is the exclusive licensee.
Please cite this article as: Martin C, Aguilo N, Gonzalo-Asensio J. Vacunación frente a tuberculosis. Enferm Infecc Microbiol Clin. 2018;36:648–656.
