






This article reviews the different acute and chronic neurological manifestations of excessive alcohol consumption that affect the central or peripheral nervous system. Several mechanisms can be implicated depending on the disorder, ranging from nutritional factors, alcohol-related toxicity, metabolic changes and immune-mediated mechanisms. Recognition and early treatment of these manifestations is essential given their association with high morbidity and significantly increased mortality.
En este artículo se revisan las distintas manifestaciones neurológicas del consumo excesivo de alcohol, que pueden ser agudas o crónicas y afectar al sistema nervioso central o periférico. El mecanismo por el cual se producen varía de un grupo de trastornos a otro. Destacan factores nutricionales, efectos tóxicos del alcohol, factores metabólicos e incluso inmunológicos. Estas manifestaciones pueden conllevar una gran morbilidad y un aumento significativo de la mortalidad, por lo que es importante reconocerlas y tratarlas precozmente.
Excessive or harmful alcohol consumption is defined as the consumption of 40–60g/day of alcohol in women or 60–100g/day in men. Although it does not meet the criteria for alcohol dependency, this level of consumption can produce clinical changes. Alcohol use disorder appears when excessive alcohol consumption causes the deterioration of an individual's social, work and family relationships.1
The World Health Organization's report on excessive alcohol consumption identified more than 60 alcohol-related diseases.2 The systemic effects of alcohol include changes in the digestive tract and the liver, the heart and vascular system, the skeletal and muscular systems, nutritional status, the immune, endocrine and haematological systems, and in the central and peripheral nervous systems.
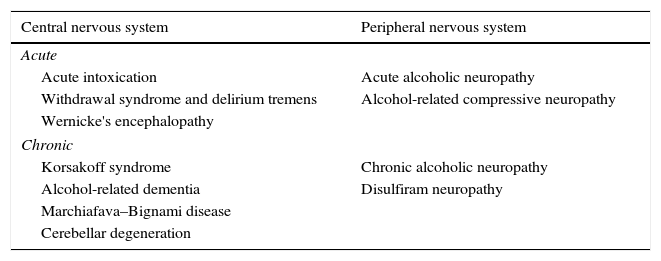
Several alcohol-related neurological complications have been described (Table 1), and pathogenesis varies greatly among the different disorder groups, although one of the most frequent causes is nutritional deficiency. Alcohol can produce alcoholic liver disease that can be accompanied by a wide variety of neurological manifestations, including hepatic encephalopathy (HE).
Neurological manifestations secondary to excessive alcohol consumption.
| Central nervous system | Peripheral nervous system |
|---|---|
| Acute | |
| Acute intoxication | Acute alcoholic neuropathy |
| Withdrawal syndrome and delirium tremens | Alcohol-related compressive neuropathy |
| Wernicke's encephalopathy | |
| Chronic | |
| Korsakoff syndrome | Chronic alcoholic neuropathy |
| Alcohol-related dementia | Disulfiram neuropathy |
| Marchiafava–Bignami disease | |
| Cerebellar degeneration | |
In this article, we will examine the different neurological manifestations of excessive alcohol consumption, the neurological alterations most frequently found in alcoholic liver disease—such as HE—and the best diagnostic approach in clinical practice.
Central nervous system involvementAcute complicationsAcute intoxicationThe symptoms of alcohol intoxication are the result of the inhibitory effect of alcohol on the nerve cells of the brain and spinal cord. Some of the immediate effects of acute alcohol ingestion—such as loquacity, loss of social inhibition, and aggressiveness—appear to be due to the inhibition of certain subcortical structures (perhaps the midbrain reticular formation) that modulate the activity of the cerebral cortex.3 However, as more alcohol is consumed, this inhibitory action extends to cortical and other brain stem and spinal neurons, and can cause decreased alertness and coma with respiratory failure. Some susceptible individuals may experience amnesic lacunae and seizures after relatively mild alcohol intoxication.4
The severity of symptoms of acute alcohol intoxication are related to blood alcohol levels5 (Table 2). These levels should be taken merely as a guide, and will vary between individuals according to sex, habitual consumption, and genetic and metabolic factors.
Manifestations of acute alcohol intoxication.
| Blood alcohol, in mg/dl | Effects |
|---|---|
| <50 | Difficulty in performing tasks that require skill, euphoria, loquacity |
| >100 | Loss of self-control, loss of coordination, mental slowness, mild dysarthria, ataxia, altered perception |
| >200 | Amnesia, confusion, diplopia, dysarthria, hypothermia, nausea, vomiting |
| >400 | Stupor, respiratory depression, coma |
Acute alcohol intoxication should be treated with supportive measures and monitoring of the individual's level of consciousness. Alcoholic coma, with its associated respiratory depression, is a medical emergency that requires appropriate life support measures.
Alcohol withdrawal syndromeAlcohol withdrawal syndrome, or abstinence syndrome, is the clinical manifestation of abruptly terminating or substantially reducing intake in patients who have developed tolerance and dependence. Alcohol acts basically through 2 specific neuronal receptors. On the one hand, it regulates the neurotransmitter gamma-aminobutyric acid type A receptor that inhibits neuronal excitability, which explains its sedative and hypnotic effects. On the other hand, alcohol increases glutamate N-methyl-d-aspartate receptor expression, which in turn increases glutamate activity and causes hyperexcitation. Chronic alcohol consumption induces neuroadaptive changes (tolerance), increases glutamate N-methyl-d-aspartate receptor expression, and desensitises the response and the expression of gamma-aminobutyric receptors.6
The manifestations of withdrawal syndrome (a wide range of severe symptoms, ranging from distal hand tremor, anxiety, insomnia and visual hallucinations to psychomotor agitation, autonomic hyperactivity, seizures or coma) appear to be mediated by an increase in excitatory neurotransmitters at the expense of inhibitory neurotransmitters. Symptoms typically onset 6–24h after interruption or reduction of alcohol consumption. The most serious form, which usually appears 72h after withdrawal, is delirium tremens, characterised by disorientation, agitation and visual hallucinations, accompanied by autonomic signs such as hyperventilation, tachycardia and diaphoresis. It can also be accompanied by metabolic and electrolyte alterations, such as hypomagnesaemia. The mortality rate is 5–15%, mainly due to metabolic, cardiovascular and infectious complications.7
According to the European Association for the Study of the Liver, the treatment of choice in patients with acute withdrawal syndrome and alcoholic liver disease is benzodiazepines,8 since they reduce the risk of epileptic seizures. Long-acting benzodiazepines, such as diazepam, are used, and the dose should be tapered over time. However, in elderly patients, in patients with hepatic failure, or when excessive sedation must be avoided, the lowest possible dose of short- or intermediate-acting benzodiazepines, such as lorazepam, is recommended. In the case of hallucinations and agitation that do not respond to benzodiazepines, haloperidol may be added, although it should only be used in combination with benzodiazepines, because administration of antipsychotics alone may increase the risk of seizures. Other drugs, such as alpha-2 agonists (clonidine and dexmedetomidine) and beta-blockers, can be used as adjuvant treatments to control autonomic hyperactivity. Studies in other drugs, such as carbamazepine, gabapentin and topiramate, have so far yielded promising results.9
Wernicke's encephalopathyWernicke's encephalopathy (WE) and Korsakoff syndrome, which were originally described as separate entities, are now considered the acute and chronic stages, respectively, of Wernicke–Korsakoff syndrome. The real prevalence of WE cannot be accurately estimated, although different studies have observed typical WE lesions in 0.2–2.8% of the autopsies performed in the general population compared with a prevalence of 12.5% in autopsies performed on alcoholics.10
WE is caused by a vitamin B1 (thiamine) deficiency, which plays a key role in carbohydrate metabolism as an essential coenzyme in the Krebs cycle and the pentose phosphate pathway (transketolase, α-ketoglutarate dehydrogenase, pyruvate dehydrogenase, etc.). Since these enzymes regulate energy metabolism in the brain, thiamine deficiency can cause brain damage, mainly in regions with greater metabolic demand, such as the paraventricular region of the thalamus and hypothalamus, the mammillary bodies, the periaqueductal grey, the floor of the fourth ventricle, and the cerebellar vermis. Thiamine is found in both animal and plant foods. It is absorbed by the duodenum, and bodily reserves can be depleted in 2–3 weeks.
In developed countries, more than 80% of cases of WE occur in the context of malnutrition associated with alcohol consumption. However, studies have shown that WE in alcoholics can involve mechanisms other than malnutrition, such as impaired gastrointestinal absorption of thiamine and a reduced capacity to store and metabolise the vitamin in the liver.11 Other clinical situations that lead to thiamine deficiency should also be borne in mind. These generally involve poor intestinal absorption (gastrointestinal surgery, hyperemesis gravidarum) or an increase in body requirement (systemic diseases).12
From the clinical point of view, WE is characterised by the classic triad of oculomotor disturbance, ataxia and confusion, although the complete triad occurs only in 16% of patients.13 Ocular alterations are complex, and consist mainly of a combination of alterations such as, for example, horizontal or vertical nystagmus, unilateral or bilateral oculomotor paresis, or conjugate gaze palsy. Ataxia mainly affects the trunk by altering gait and balance; limb ataxia and dysarthria are less common. Confusion or encephalopathic symptoms, meanwhile, develop within days or weeks and are characterised by profound disorientation, inability to concentrate, apathy, indifference, inattention, drowsiness and coma. Other signs and symptoms are hypothermia resulting from posterior hypothalamic involvement, tachycardia or postural hypotension due to autonomic nervous system dysfunction, or polyneuropathy due to multiple vitamin deficiency.12
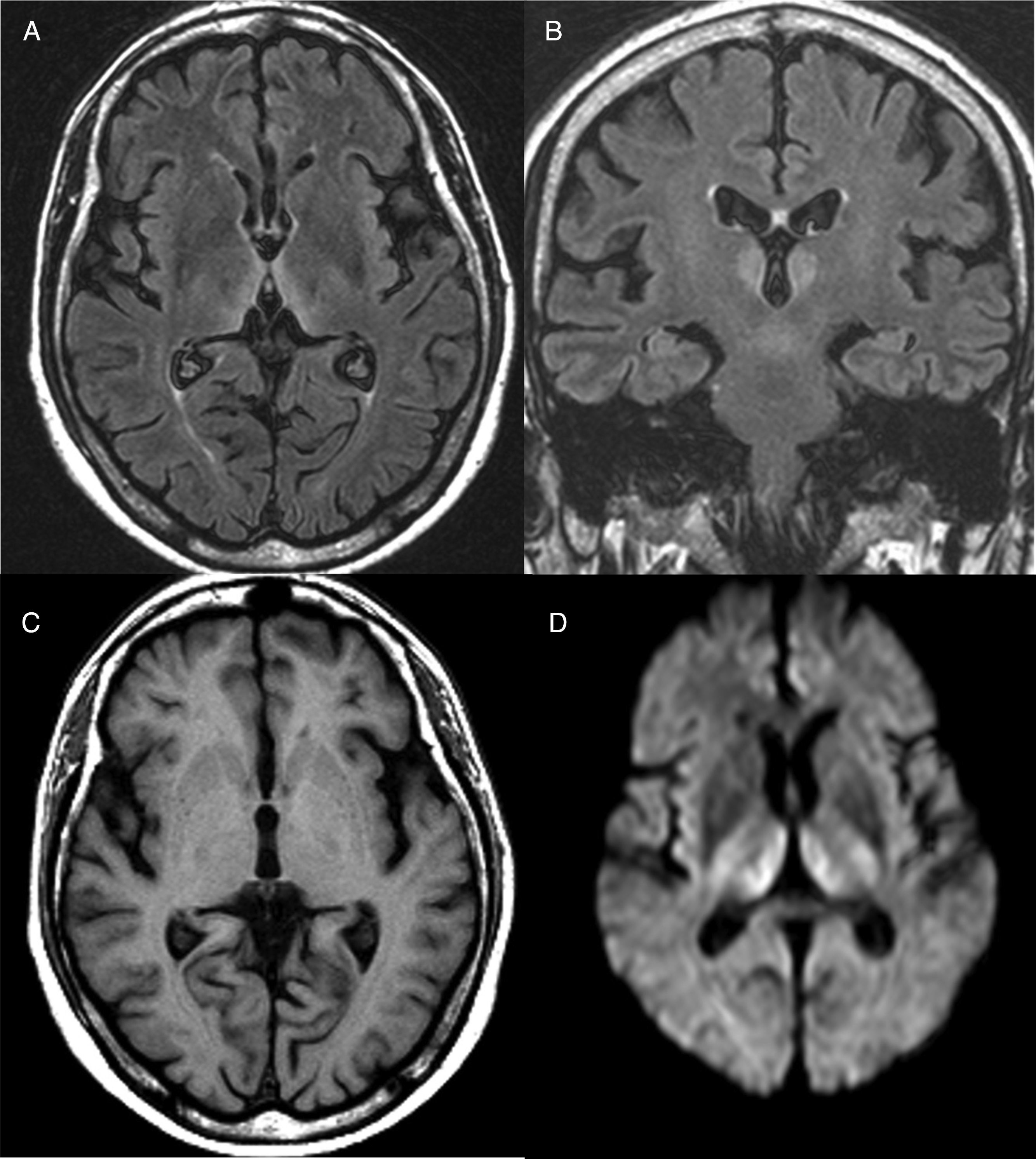
Diagnosis is mainly clinical. Some additional tests can help confirm or rule out other suspicions, but should never delay the start of treatment. A thiamine blood test will show thiamine serum levels and transketolase enzyme activity in peripheral blood. This test, however, usually takes time and is of little practical use since normal ranges do not rule out a diagnosis of WE. As far as brain imaging tests are concerned, the most useful complementary test for confirming diagnosis is magnetic resonance imaging (MRI). The most distinctive lesion is reversible cytotoxic oedema, visualised in T2, FLAIR and DWI sequences in the periventricular region and diencephalon (Fig. 1). Furthermore, mammalian body atrophy, which is usually present in patients with chronic lesions, can start to be detected within the first week after onset of the disease.14

Brain magnetic resonance image from a patient with Wernicke's encephalopathy. Bilateral thalamic hyperintensities around the third ventricle are seen on fluid attenuation inversion recovery (FLAIR) weighted axial (A) and coronal (B) images. (C) T1-weighted axial image showing no lesions. (D) Diffusion-weighted (DWI) axial image showing hyperintensity in bilateral symmetric thalami.
WE is a medical emergency because it is potentially reversible and delayed treatment or no treatment at all can cause serious sequelae and even death.
Treatment consists of urgent thiamine replacement. Thiamine therapy has been evaluated in a single randomised double-blind study in 107 patients, in which the efficacy of different intramuscular doses (5, 20, 50, 100 and 200mg) of thiamine daily for 2 days was compared. Response, defined as improved neuropsychological test score, was evaluated on the third day after treatment. The authors concluded that the 200mg dose was superior to the other dosages.15 Although there is no clear consensus on the ideal thiamine dose, pharmacokinetic studies have shown the half-life of thiamine to be around 96min, so 2 or 3 daily doses are considered appropriate.16 Given the higher incidence of adverse effects in intramuscular administration (high volume and painful administration), intravenous infusion of thiamine diluted in 100ml of physiological saline or 5% dextrose over 30min is recommended. According to evidence from published series and the recommendations of the European Federation of Neurological Societies, the administration of between 100 and 200mg of intravenous thiamine is considered adequate in non-alcoholic patients, while alcoholic patients require doses of up to 500mg 3 times daily.10 Other recommendations are a speedy return to a normal diet and continuous treatment until clinical improvement is observed. According to the literature, when untreated or insufficiently treated, WE-induced brain damage can lead to death in 20% of cases or to the chronic form of WE (Korsakoff syndrome) in 80% of cases.17
Chronic complicationsKorsakoff syndromeKorsakoff syndrome, which is mainly caused by malnutrition associated with chronic alcoholism, usually emerges in the aftermath of WE, although it can sometimes appear in patients with no history of WE, or with sub-acute, undiagnosed episodes. However, it can also be a symptom of malnutrition due to other causes or a symptom of diseases involving ischaemic, neoplastic or other injury to the medial and inferomedial thalamic regions in the temporal lobes.18
From a clinical point of view, it is characterised by memory impairment that is out of proportion to other cognitive functions in an awake, attentive and responsive patient. Important manifestations are learning deficits and memory loss, affecting both anterograde and retrograde events. Recent memory is usually more affected than remote memory, and the creation of false memories, or confabulation, in speech that can even be induced by questions about the patient's recent activities is characteristic of this syndrome. Other cognitive functions, such as concentration, spatial organisation, visual or verbal abstraction, may also be affected, and patients are usually apathetic and lacking in initiative, spontaneity and self-criticism.19
Korsakoff syndrome is often thought to be untreatable; however, after thiamine administration, only 25% show no recovery, 25% experience discrete improvement, 25% significant improvement, and in 25% memory is completely recovered.20
Marchiafava–Bignami diseaseMarchiafava–Bignami disease was first described in 1903 in Italian alcoholic wine drinkers, and since then it has been observed in other nationalities in association with abuse of any alcoholic beverage. It affects chronic alcoholics almost exclusively, although cases have occasionally been described in non-alcoholics with malnutrition.21 It is a rare entity that is characterised by progressive demyelination and necrosis of the central part of the corpus callosum. On brain imaging studies, it manifests as a well defined area of demyelination in the body of the corpus callosum, which can then extend to various regions of the subcortical white matter.
The aetiology is unknown and widely debated, with some experts suggesting the existence of a toxic factor, not yet identified, which is present in some alcoholic beverages as the culprit. However, given the low prevalence of this entity in alcoholics, and the fact that it has also been described in some non-alcoholics, it probably has an unidentified nutritional, metabolic or enzymatic aetiology.22
The clinical characteristics of this disease vary, and there is no well-defined clinical syndrome. Most patients present progressive dementia, usually subacute at onset, with predominance of apractic or aphasic disorders, oppositional hypertonia, dysarthria, frontal release reflexes and, sometimes, hemiparesis or signs of interhemispheric disconnection. Some patients also present decreased alertness and seizures. The clinical course is variable; some patients will become comatose and die, others can survive several years with dementia, while in others partial recovery is possible.23
The diagnosis is difficult because of the wide spectrum of symptoms. Diagnosis was hitherto based on post-mortem studies, but today a history of alcoholism together with clinical manifestation and, above all, brain imaging studies, specifically MRI, are essential to confirm diagnosis. Typical lesions seen on MRI are demyelination, swelling and necrosis of the corpus callosum, with varying degrees of subcortical white matter involvement.24
Given its uncertain aetiology, there is no specific treatment for Marchiafava–Bignami disease, although abstinence and vitamin supplements are recommended. Good response to high doses of corticosteroids has also been reported in some studies.25
Alcohol-related dementiaThe term alcohol-related dementia is used to describe a form of dementia attributable to the direct effects of chronic alcohol consumption on the brain. Studies have shown that consumption of 140g or more of alcohol per day for a prolonged period of time can produce moderate cognitive alterations.26
However, alcohol-related dementia has never been accurately defined from a clinical or pathological perspective, and the diagnosis of this entity has sparked considerable controversy in recent years. Furthermore, the interaction between nutritional deficiencies, consumption of other substances, psychiatric comorbidity and repeated head injuries in chronic alcoholic patients raise doubts about the existence of alcohol-related dementia per se. Some authors, therefore, prefer to use the term “alcohol-related brain damage” to describe both the aetiology and symptoms of the widely differing alcohol-related cognitive disorders presented in these patients.
Neuronal damage or depletion, from a physiopathological perspective, is believed to be related to glutamate neurotoxicity, oxidative stress and diminished neurogenesis, triggered by chronic alcohol abuse.27
Post-mortem anatomopathological studies of patients diagnosed with alcohol-related dementia often show nonspecific findings, such as predominantly frontal cerebral atrophy, typical Wernicke–Korsakoff syndrome lesions, communicating hydrocephalus, Alzheimer's disease, or traumatic injuries of variable severity.28
From a clinical point of view, it is characterised by an insidious onset with stepwise progression of symptoms that overlap with other neurodegenerative dementias. In the early stages, neuropsychological studies usually reveal frontal-subcortical cognitive impairment, with slowing of mental processes, attention deficit, changes in immediate or short-term memory, decline in visual spatial skills, and decline in executive functions, such as planning and organisation.29
Alcohol-related cerebellar degenerationAlcohol-related cerebellar degeneration is a common complication that affects up to 25% of alcoholics, and is one of the most frequent causes of acquired ataxia in adults.30
The pathogenesis of this entity is complex and not entirely clear, although a synergistic mechanism involving both the toxic effect of alcohol and the consequences of vitamin B1 deficiency may be involved. Recent studies have shown the presence of anti-tissue transglutaminase 2 antibodies in chronic alcoholics, which raises the possibility of alcohol-induced hypersensitivity to gluten.31 According to the hypothesis put forward by some authors to explain this phenomenon, increased gut permeability caused by alcohol-induced intestinal mucosa lesions found in alcoholic patients could increase exposure of the immune system to pathogenic antigens (including gliadin peptides). Then, blood–brain barrier impairment induced by chronic alcohol consumption would, by as yet unknown mechanisms, allow the passage of these antibodies to the brain, thus causing cerebellar degeneration similar to gluten-induced cerebellar ataxia.32 In fact, one study has shown a higher prevalence of anti-gliadin antibodies in patients with alcohol-induced cerebellar degeneration compared to the general population (44% vs 12%).33
From a clinical perspective, cerebellar degeneration is characterised by trunk ataxia with wide-based gait, instability and variable degrees of lower limb dysmetria. Dysmetria in the upper limbs, dysarthria or oculomotor disturbance are less common. In most cases, cerebellar degeneration evolves over a period of several weeks or months and persists for years.
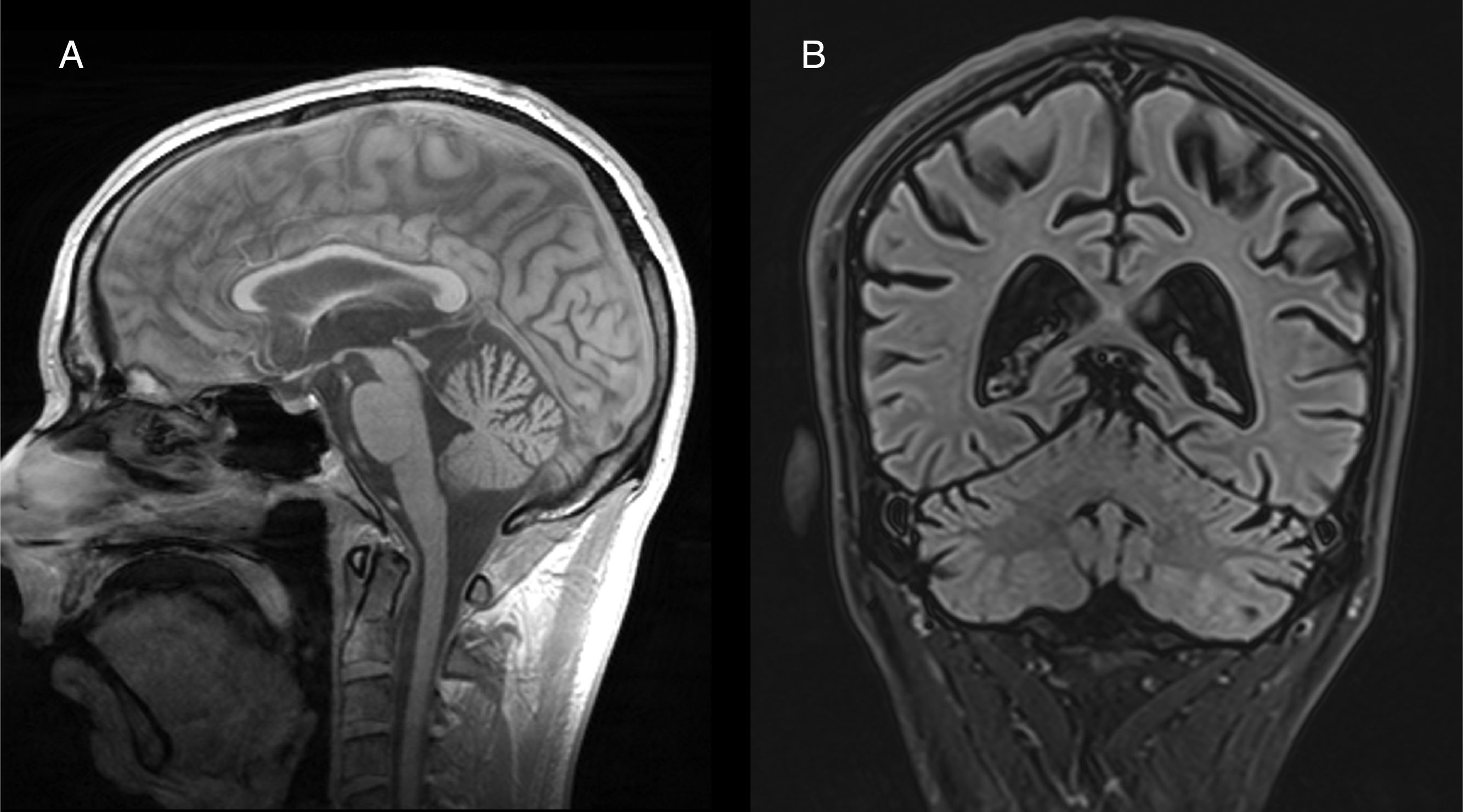
Both anatomopathological and neuroimaging studies show degeneration of all the neurocellular elements of the cerebellar cortex, and particularly Purkinje cells in the anterior and superior surface of the vermis.34 Cerebellar atrophy is easily observed on CT and brain MRI scans (Fig. 2). No specific treatment has been defined, although administration of vitamin supplements and abstinence from alcohol are recommended.
Peripheral nervous system involvementAcute compressive neuropathy sagittal and (B) coronal images showing predominantly cerebellar vermian atrophy.")
Excessive alcohol consumption is traditionally associated with ‘Saturday night palsy’, caused by compression of the radial nerve against the humerus for several hours. It usually occurs when the individual falls asleep with their arm hanging over the armrest of a chair, or being compressed by the weight of the body. Clinically, it is characterised by an inability to perform dorsiflexion of the wrist and extension of the fingers. Neurophysiological studies using electromyography is useful both for diagnosis and prognosis, although the condition is self-limiting and most patients recover within 3–6 months.35
Chronic alcoholic polyneuropathyChronic polyneuropathy is the most common complication in alcoholic patients.36 Recently, strong evidence has come to light to show alcoholic polyneuropathy to be the result of a multifactorial process primarily mediated by the toxic effect of alcohol and modulated by other factors, such as genetic predisposition, thiamine deficiency, malnutrition and other systemic diseases.37
This is a predominantly axonal, sensorimotor polyneuropathy with distal, symmetric features. The onset of symptoms is insidious and symmetric, predominantly sensory, in the form of dysesthesia, burning sensation and burning pain on the soles of the feet that later develops into cramp in the calves and the hands. Motor symptoms usually manifest later, and are characterised by muscle weakness and atrophy, especially in the distal muscles of the upper or lower limbs. Vegetative vascular and skin defects (sweaty, atrophic, glossy, almost hairless skin) with associated dysautonomia are also typical.
The treatment consists of a balanced diet with vitamin supplements, rehabilitation and alcohol abstinence. However, recovery is slow and often incomplete. Patients presenting with neuropathic pain can be treated with drugs such as gabapentin or amitriptyline.
Disulfiram neuropathyDisulfiram, a drug used to facilitate alcohol abstinence, has very occasionally been associated with peripheral neuropathy. The physiopathological mechanism behind this is unknown, but it manifests as an axonal sensorimotor, dose-dependent polyneuropathy that onsets a few weeks or months after the start of disulfiram treatment. Clinically, it is characterised by distal paresthesias in a stocking-glove distribution together with predominantly distal muscle weakness and distal areflexia. Prognosis will depend on severity and the degree of axonal loss, although it is usually reversible after suspension of disulfiram therapy.38
Muscle involvementAcute or chronic alcoholic myopathyAlcohol can damage skeletal muscles by altering calcium channels or the integrity of the muscle fibre membrane or sarcolemma. Clinically, alcoholic myopathy can be characterised by acute myalgia, rhabdomyolysis and elevated creatine kinase (CK). In severe cases, it can lead to acute kidney failure and myoglobinuria. The chronic form presents as muscle atrophy, usually proximal, which frequently coexists with neuroperipheral alterations, such as chronic alcoholic polyneuropathy. Diagnosis is performed with neurophysiological tests (electromyography) and histopathological study of muscle biopsy. In some cases, skeletal myopathy is accompanied by dilated cardiomyopathy. Treatment is based on alcohol abstinence, physiotherapy and a balanced diet. Prognosis is cautious, since some patients experience an improvement in clinical weakness, but others do not recover muscle strength or mass.39
Hepatic encephalopathyHepatic encephalopathy (HE) is a common, serious complication of liver cirrhosis and an indicator of poor prognosis in these patients. It is a complex syndrome involving neurological and psychiatric manifestations, and occurs in patients with advanced liver disease and portosystemic shunting.40
Symptoms fluctuate over time and vary greatly, ranging from tremor and dysarthria to hepatic coma, and include (a) altered level of consciousness that can progress from mild confusion to coma; (b) neuropsychiatric symptoms, such as behavioural changes, mental slowness, reversal of the sleep–wake cycle, or psychomotor agitation, and (c) neuromuscular symptoms, such as flapping. The West Haven criteria stratifies HE into 4 grades, from mild to most severe (Table 3). Because the clinical manifestations of HE are non-specific and can be observed in other diseases or metabolic disorders, diagnosis is made on the basis of complementary tests that can reasonably exclude other potential causes.
West Haven criteria for hepatic encephalopathy.
| Grades of hepatic encephalopathy |
|---|
| I Euphoria, anxiety, shortened attention span, reversal of sleep–wake cycle. Sporadic flapping |
| II Lethargy, apathy, disorientation in time and space, behaviour disorder. Evident flapping |
| III Deep somnolence, stupor, confusional state, inappropriate behaviour, gross disorientation. Flapping at times impossible to evaluate due to lack of collaboration |
| IV Coma |
Diagnosis of WE in patients with chronic alcoholism is challenging, since the differential diagnosis is extensive.
Clinically, the diagnosis of WE can be difficult because, as explained previously, the classic triad is often absent. The presence of alterations in ocular motility, nystagmus and ataxia should raise suspicion of WE, while the presence of flapping or pyramidal syndrome is suggestive of HE. Clinical findings suggestive of alcohol withdrawal are anxiety, tachycardia, visual hallucinations and postural tremor. Differential diagnosis between HE and Marchiafava–Bignami disease does not generally present any difficulty, since the latter is clinically characterised by the presence of dementia and spasticity.
Brain neuroimaging using computed tomography (CT) or MRI makes it possible to rule out structural alterations, such as space-occupying lesions, subdural haematomas or ischaemic/haemorrhagic strokes, which can be suspected when neurological focal signs are observed in the examination. These imaging studies can also rule out viral or autoimmune encephalitis that can clinically overlap HE. A typical MRI finding in HE is hyperintense basal ganglia on T1-weighted images, especially in the globus pallidus. This is related to manganese deposits caused by portosystemic shunts, and could explain the existence of parkinsonian signs in these patients.41
An electroencephalogram, a neurophysiological test that shows the electrical activity of the brain, can rule out a non-convulsive status epilepticus or findings typical of a post-critical state. However, HE produces alterations in brain activity that are shown on the electroencephalogram as slow frequency, high amplitude waves and three-phase waves—findings that can also be found in other metabolic comas.42
Finally, the study of cerebrospinal fluid will rule out meningitis or bacterial or viral meningoencephalitis, which should be suspected when fever and meningeal signs are present.
ConclusionsChronic alcohol consumption can produce numerous neurological manifestations. The most common are polyneuropathy, cerebellar degeneration and dementia, and the most serious are WE, Korsakoff syndrome and Marchiafava–Bignami disease. All these are associated with significant morbidity and mortality, and therefore must be correctly diagnosed and treated in order to avoid irreversible complications. They are caused by vitamin deficiencies, the direct toxic effects of alcohol, immune alterations and unknown mechanisms, among others. In addition, the differential diagnosis of HE can be difficult due to its similarity with alcohol-related neurological disorders.
Conflicts of interestThe authors declare that they have no conflicts of interest.
We would like to thank the Institute of Diagnostic Imaging (IDI), Magnetic Resonance Unit, Hospital Germans Trias i Pujol (Badalona, Barcelona, Spain), and particularly Fidel Núñez.
Please cite this article as: Planas-Ballvé A, Grau-López L, Morillas RM, Planas R. Manifestaciones neurológicas del alcoholismo. Gastroenterol Hepatol. 2017;40:709–717.
