






En este artículo se revisan las distintas manifestaciones neurológicas del consumo excesivo de alcohol, que pueden ser agudas o crónicas y afectar al sistema nervioso central o periférico. El mecanismo por el cual se producen varía de un grupo de trastornos a otro. Destacan factores nutricionales, efectos tóxicos del alcohol, factores metabólicos e incluso inmunológicos. Estas manifestaciones pueden conllevar una gran morbilidad y un aumento significativo de la mortalidad, por lo que es importante reconocerlas y tratarlas precozmente.
This article reviews the different acute and chronic neurological manifestations of excessive alcohol consumption that affect the central or peripheral nervous system. Several mechanisms can be implicated depending on the disorder, ranging from nutritional factors, alcohol-related toxicity, metabolic changes and immune-mediated mechanisms. Recognition and early treatment of these manifestations is essential given their association with high morbidity and significantly increased mortality.
El consumo excesivo o perjudicial de alcohol es aquel que, sin cumplir criterios de dependencia, puede producir alteraciones médicas en un individuo y se define como el consumo de alcohol de 40-60 g/día en mujeres o de 60-100 g/día en hombres. El trastorno por dependencia de alcohol aparece cuando el consumo excesivo de esta sustancia produce un deterioro en las relaciones sociales, laborales y familiares del individuo1.
El informe de la Organización Mundial de la Salud sobre el consumo excesivo de alcohol identificó más de 60 enfermedades relacionadas2. Los efectos sistémicos del alcohol incluyen alteraciones sobre el tracto digestivo y el hígado, el corazón y sistema vascular, los sistemas óseo y muscular, el estado nutricional, los sistemas inmunológico, endocrinológico y hematológico y sobre los sistemas nerviosos central y periférico.
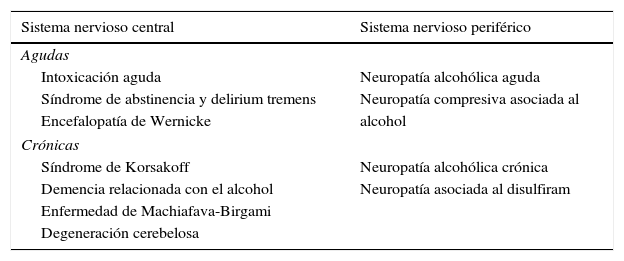
Las complicaciones neurológicas relacionadas con el alcoholismo son numerosas (tabla 1) y el mecanismo por el que se producen varía de manera notable de un grupo de trastornos a otro, aunque uno de los más frecuentes es la carencia nutricional. Asimismo, el alcohol puede producir una enfermedad hepática alcohólica que puede acompañarse de una amplia variedad de manifestaciones neurológicas, entre las que destaca la encefalopatía hepática (EH).
Manifestaciones neurológicas secundarias al consumo excesivo de alcohol
| Sistema nervioso central | Sistema nervioso periférico |
|---|---|
| Agudas | |
| Intoxicación aguda | Neuropatía alcohólica aguda |
| Síndrome de abstinencia y delirium tremens | Neuropatía compresiva asociada al |
| Encefalopatía de Wernicke | alcohol |
| Crónicas | |
| Síndrome de Korsakoff | Neuropatía alcohólica crónica |
| Demencia relacionada con el alcohol | Neuropatía asociada al disulfiram |
| Enfermedad de Machiafava-Birgami | |
| Degeneración cerebelosa | |
En este artículo se examinan las distintas manifestaciones neurológicas del consumo excesivo de alcohol, las alteraciones neurológicas más frecuentes en la hepatopatía alcohólica —como la EH— y la sistemática más conveniente para reconocerlas en la práctica clínica.
Afectación del sistema nervioso centralComplicaciones agudasIntoxicación agudaLos síntomas de la intoxicación alcohólica son resultado de la acción depresora del alcohol sobre las neuronas cerebrales y espinales. Al parecer, algunos de los efectos inmediatos de la ingestión aguda de alcohol —como locuacidad, pérdida de inhibición social y agresividad— podrían ser debidos a la inhibición de ciertas estructuras subcorticales (tal vez la formación reticular de la parte alta del tallo cerebral) que modulan la actividad de la corteza cerebral3. Sin embargo, con cantidades crecientes de alcohol la acción depresora se extiende para abarcar también las neuronas corticales y otras del tallo cerebral y espinales, de manera que puede llegar a producirse una disminución del nivel de conciencia y un coma con depresión respiratoria. Las lagunas amnésicas y las crisis convulsivas pueden aparecer con facilidad en algunas personas susceptibles tras intoxicaciones relativamente leves4.
Existe una relación entre los síntomas derivados del consumo agudo de alcohol y los niveles en sangre5 (tabla 2). Estas cifras deben tomarse como orientativas, ya que hay variaciones interindividuales en función del sexo, consumo habitual, factores genéticos y metabólicos.
Manifestaciones de la intoxicación aguda alcohólica
| Alcoholemia en mg/dl | Efectos |
|---|---|
| <50 | Alteración en tareas que requieren habilidad, euforia, locuacidad |
| >100 | Pérdida del autocontrol, incoordinación, lentitud mental, disartria leve, ataxia, alteración de la percepción |
| >200 | Amnesia, confusión, diplopía, disartria, hipotermia, náuseas, vómitos |
| >400 | Estupor, depresión respiratoria, coma |
El tratamiento de la intoxicación alcohólica aguda es de soporte y de vigilancia del nivel de conciencia. El coma alcohólico representa una urgencia médica —sobre todo debido a la depresión de la función respiratoria— que requiere medidas apropiadas de soporte vital.
Síndrome de privación alcohólicaEl síndrome de privación alcohólica o síndrome de abstinencia es la expresión clínica de la interrupción brusca o disminución de la ingestión de alcohol en aquellos pacientes que han desarrollado tolerancia y dependencia. El alcohol actúa básicamente a través de 2receptores neuronales específicos. Por un lado, regula el receptor de tipo A del ácido gamma-aminobutírico, que produce una menor excitabilidad neuronal, lo que explica sus efectos sedantes e hipnóticos. Por otro lado, el alcohol aumenta el número de receptores para glutamato de N-metil-D-aspartato, lo que conlleva un aumento de la función glutamatérgica, con el consiguiente estado de hiperexcitación. El consumo crónico de alcohol induce cambios neuroadaptativos (tolerancia), aumenta el número de los receptores para el glutamato N-metil-D-aspartato y desensibiliza la respuesta y el número de receptores gamma-aminobutíricos6.
Las manifestaciones del síndrome de abstinencia (una amplia gama de gravedad, desde temblor distal de manos, ansiedad, insomnio y alucinaciones visuales hasta agitación psicomotora, hiperactividad autonómica, crisis convulsivas o coma) parecen estar mediadas por un aumento en la neurotransmisión excitatoria con respecto a la inhibitoria. Los síntomas empiezan típicamente tras 6-24 h después de la interrupción o disminución del consumo de alcohol. La forma más grave, que suele aparecer a las 72 h de la privación, es el delirium tremens, que se caracteriza por desorientación, agitación y alucinaciones visuales, acompañado de signos autonómicos como hiperventilación, taquicardia y diaforesis. Puede asociar alteraciones metabólicas e hidroelectrolíticas como la hipomagnesemia. La mortalidad es del 5 al 15%, fundamentalmente por complicaciones metabólicas, cardiovasculares e infecciosas7.
Según la Asociación Europea para el Estudio del Hígado, en los pacientes con síndrome de abstinencia agudo y hepatopatía alcohólica el tratamiento de elección son las benzodiacepinas8, ya que disminuyen el riesgo de crisis epilépticas. Se utilizan las de vida media prolongada, como el diacepam, y es aconsejable reducir la dosis gradualmente. Sin embargo, en pacientes ancianos, en pacientes con insuficiencia hepática, o cuando quiere evitarse una sedación excesiva, son recomendables las benzodiacepinas de vida media corta o intermedia, como el lorazepam a las menores dosis posibles. Si el paciente presenta alucinaciones y agitación que no responden a benzodiacepinas, puede añadirse haloperidol, aunque debe usarse solo en combinación con benzodiacepinas debido a que los antipsicóticos aislados pueden aumentar el riesgo de convulsiones. Otros fármacos como agonistas alfa-2 (clonidina y dexmetomidina) y betabloqueantes pueden utilizarse como tratamientos adyuvantes para controlar la hiperactividad autonómica. Se están investigando otros medicamentos con resultados prometedores como la carbamacepina, gabapentina y el topiramato9.
Encefalopatía de WernickeEl síndrome de Wernicke-Korsakoff incluye la encefalopatía de Wernicke (EW) y el síndrome amnésico de Korsakoff. Aunque históricamente han sido descritas como entidades separadas, hoy día se considera que son la fase aguda y el estado residual, respectivamente, del mismo proceso patológico. La prevalencia real no puede establecerse con precisión, aunque diferentes estudios han observado una prevalencia de lesiones típicas de EW entre el 0,2 y el 2,8% de las autopsias realizadas en la población general, y del 12,5% en autopsias realizadas a alcohólicos10.
Esta entidad está causada por el déficit de vitamina B1 (tiamina), la cual tiene un papel fundamental en el metabolismo de los hidratos de carbono como cofactor de enzimas esenciales para el ciclo de Krebs y la vía pentosa fosfato (transcetolasa, α-cetoglutarato-deshidrogenasa, piruvato-deshidrogenasa…). Dado que dichas enzimas regulan el metabolismo energético cerebral, el déficit de tiamina puede provocar daños, principalmente en aquellas regiones con mayor demanda metabólica como son la región paraventricular del tálamo e hipotálamo, los cuerpos mamilares, las áreas periacueductales, el suelo del IV ventrículo y el vermis cerebeloso. La tiamina está presente tanto en alimentos animales como vegetales, se absorbe por el duodeno y son necesarias 2-3 semanas para agotar las reservas del organismo.
En países desarrollados, más del 80% de los casos ocurren en contextos de malnutrición asociada al consumo de alcohol. Sin embargo, en alcohólicos, se ha demostrado que podrían estar implicados otros mecanismos aparte de la malnutrición, como son una reducción de la absorción gastrointestinal de tiamina y una disminución en la capacidad para almacenar la vitamina en el hígado y transformarla en su forma activa11. Además, es importante recordar que existen otras situaciones clínicas que también ocasionan un déficit de tiamina, generalmente con relación a un descenso en su absorción intestinal (cirugía gastrointestinal, hiperemesis gravídica) o a un aumento del requerimiento corporal (enfermedades sistémicas)12.
Desde el punto de vista clínico, la EW se caracteriza por la tríada clásica de alteración oculomotora, ataxia y confusión, si bien la tríada completa se presenta solo en un 16% de los pacientes13. Las alteraciones oculares son complejas y consisten principalmente en una combinación de alteraciones como, por ejemplo, nistagmo —que puede ser horizontal o vertical—, paresias oculomotoras uni- o bilaterales o alteraciones de la mirada conjugada. La ataxia afecta predominantemente al tronco, al provocar una alteración de la marcha y del equilibrio; es menos frecuente la ataxia de extremidades y la disartria. En cuanto al cuadro confusional o encefalopático, se desarrolla en días o semanas y se caracteriza por una profunda desorientación, dificultad para la concentración, apatía, indiferencia, inatención, somnolencia y coma. Otros síntomas y signos son la hipotermia por afectación de estructuras hipotalámicas, la taquicardia o hipotensión postural por disfunción del sistema nervioso vegetativo o la polineuropatía multicarencial12.
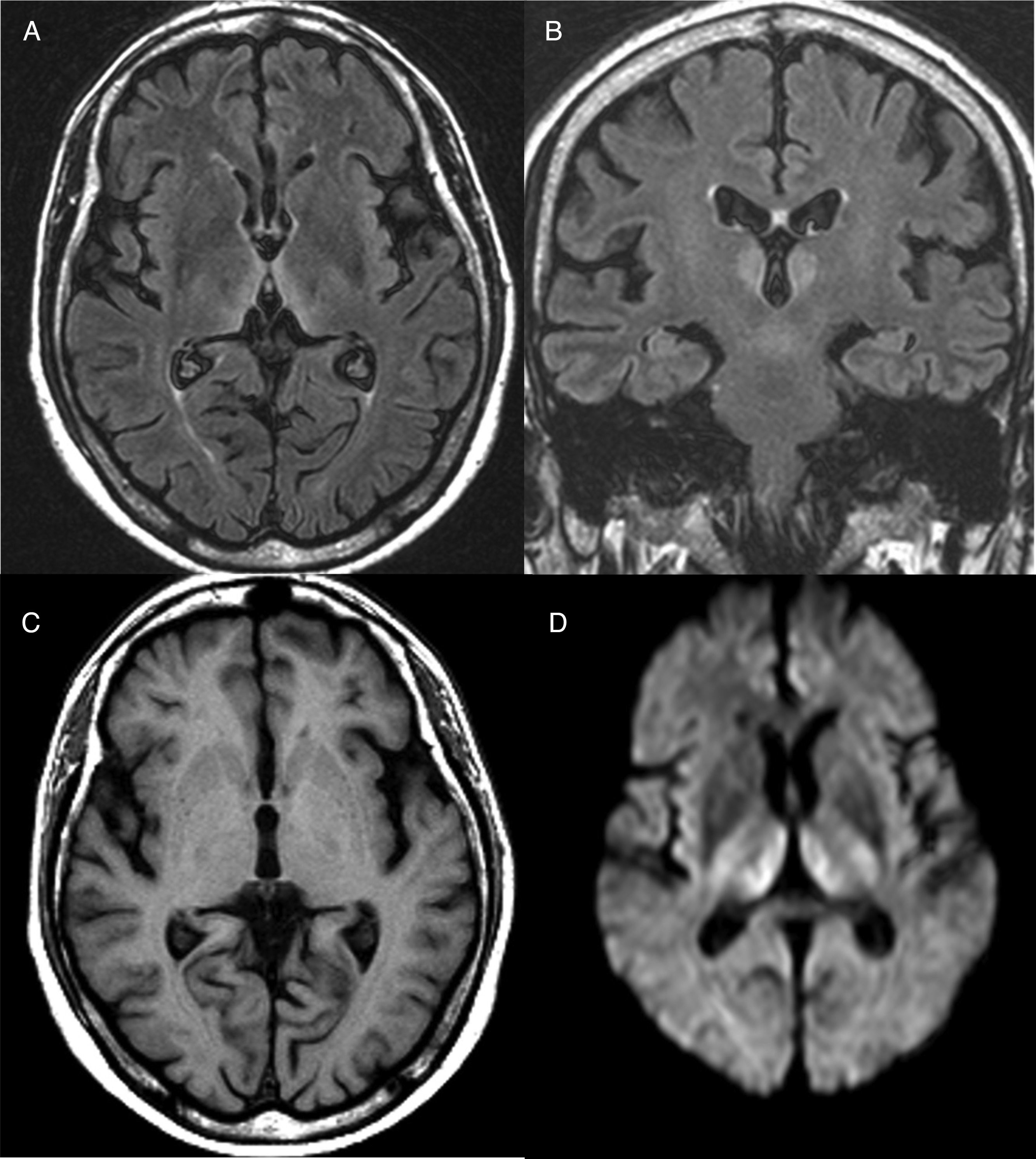
El diagnóstico es fundamentalmente clínico, aunque algunas exploraciones complementarias pueden ayudar a confirmarlo o a excluir otras alternativas diagnósticas. En ningún caso, estas exploraciones deberán retrasar el inicio del tratamiento. En sangre periférica, se pueden determinar los niveles séricos de tiamina y la actividad de la enzima transcetolasa en los hematíes, aunque son pruebas que normalmente no están disponibles de forma urgente y no son muy prácticas, dado que la normalidad de sus niveles no excluye el diagnóstico. Respecto a las pruebas de imagen cerebral, la resonancia magnética (RM) es la prueba complementaria más útil para confirmar el diagnóstico. La lesión más distintiva es el edema citotóxico reversible visualizado en secuencias T2, FLAIR y DWI en regiones periventriculares y diencefálicas (fig. 1). Además, la atrofia de los cuerpos mamilares, que suele ser una anomalía en pacientes con lesiones crónicas, puede comenzar a detectarse dentro de la primera semana del inicio de la enfermedad14.
 y coronal (B) potenciado en fluid attenuation inversion recovery (FLAIR), se aprecia hiperintensidad bitalámica medial rodeando el tercer ventrículo. C) Corte axial potenciado en T1 en el que no se observan lesiones. D) Corte axial en difusión (DWI) en el que se aprecia una hiperseñal de la difusión en ambos tálamos simétrica.")
Resonancia magnética cerebral de un paciente con encefalopatía de Wernicke. Sobre corte axial (A) y coronal (B) potenciado en fluid attenuation inversion recovery (FLAIR), se aprecia hiperintensidad bitalámica medial rodeando el tercer ventrículo. C) Corte axial potenciado en T1 en el que no se observan lesiones. D) Corte axial en difusión (DWI) en el que se aprecia una hiperseñal de la difusión en ambos tálamos simétrica.
La EW es una urgencia médica, ya que es una enfermedad potencialmente reversible y la ausencia de tratamiento o su retraso puede provocar secuelas graves e incluso la muerte.
El tratamiento consiste en la reposición de tiamina lo antes posible. La eficacia de la tiamina ha sido evaluada en un único estudio doble ciego aleatorizado con 107 pacientes en el que se comparó la eficacia de la administración de dosis de 5, 20, 50, 100 y 200mg intramusculares diariamente durante 2días, valorando como respuesta la mejoría en un test neuropsicológico realizado al tercer día tras el tratamiento. Los autores concluyeron que la dosis de 200mg era superior al resto de las dosificaciones15. A pesar de que no hay un claro consenso acerca de la posología del tratamiento con tiamina, los estudios de farmacocinética concluyen que su vida media es de unos 96 min, por lo que se considera apropiado administrarla en 2o 3dosis diarias16. Dada la mayor frecuencia de efectos adversos en la administración intramuscular (volumen elevado y administración dolorosa), se recomienda la infusión intravenosa de tiamina diluida en 100ml de suero fisiológico o glucosado al 5% en perfusión durante 30 min. De acuerdo con los datos que arroja la literatura y las recomendaciones de la Federación Europea de Sociedades de Neurología, se considera adecuada la administración de dosis de 100 a 200mg de tiamina intravenosa en pacientes no alcohólicos, mientras que los pacientes alcohólicos precisarían dosis de hasta 500mg 3veces al día10. Además, también se recomienda instaurar una dieta normal lo antes posible y continuar el tratamiento hasta la mejoría clínica. Según la experiencia reportada en la literatura, en los pacientes sin tratamiento o con tratamiento insuficiente, el daño cerebral puede llevar a la muerte en el 20% de los casos o a su forma crónica (síndrome de Korsakoff) en el 80% de los casos17.
Complicaciones crónicasSíndrome de KorsakoffEl síndrome de Korsakoff se relaciona fundamentalmente con la malnutrición asociada al alcoholismo crónico, como un estado residual de la EW, aunque en ocasiones puede aparecer sin este antecedente o tras episodios subagudos no diagnosticados. No obstante, también puede ser un síntoma de malnutrición de otras causas o síntoma de enfermedades con lesiones en regiones talámica medial o inferomedial de los lóbulos temporales, de etiología isquémica, tumoral u otras18.
Desde un punto de vista clínico, se caracteriza por un deterioro desproporcionado de la memoria con relación a otras funciones cognitivas, en un sujeto despierto, atento y con capacidad para responder. Destaca un defecto del aprendizaje y pérdida de los recuerdos, con afectación tanto de la memoria anterógrada como retrógrada. En general, la memoria reciente se encuentra más afectada que la remota y es característica la confabulación o falsificación creativa de la memoria en el discurso, que se puede incluso inducir mediante preguntas acerca de las actividades recientes del paciente. Otras funciones cognitivas como la capacidad de concentración, organización espacial, abstracción visual o verbal también pueden estar alteradas. Además, el paciente suele estar apático y carece de iniciativa, espontaneidad y autocrítica19.
El síndrome de Korsakoff a menudo se percibe como intratable, pero, de hecho, tras el tratamiento con tiamina solo el 25% no muestran ninguna recuperación, el 25% experimentan mejoría discreta, el 25% mejoría significativa y el 25% recuperan completamente la memoria20.
Enfermedad de Machiafava-BignamiLa enfermedad de Machiafava-Bignami fue descrita originalmente en el 1903 en pacientes italianos alcohólicos, bebedores de vino, y desde entonces se ha observado en otras nacionalidades y con el abuso de cualquier tipo de bebida alcohólica. Afecta casi exclusivamente a pacientes alcohólicos crónicos, aunque se han descrito casos esporádicos en abstemios con desnutrición21. Es una entidad rara que se caracteriza por una desmielinización progresiva y necrosis de la parte central del cuerpo calloso. En el ámbito neuropatológico se observa una zona de desmielinización, bien delimitada, de localización medial del cuerpo calloso, que puede extenderse a la sustancia blanca subcortical de manera variable.
La etiología es desconocida y controvertida, y se ha propuesto la existencia de un factor tóxico, todavía no identificado, presente en algunas bebidas alcohólicas. Sin embargo, dada la baja prevalencia de esta entidad en alcohólicos y el hecho de que se ha descrito también en algunos individuos abstemios, se propone una etiología nutricional, metabólica o enzimática todavía desconocida22.
En cuanto a las características clínicas, son variables y no hay un síndrome clínico bien definido. En la mayoría de los pacientes destaca una demencia progresiva de comienzo, en general, subagudo, con predominio de trastornos aprácticos o afásicos, hipertonía oposicional, disartria, reflejos de liberación frontal y, a veces, hemiparesia o signos de desconexión interhemisférica. En ocasiones también destaca alteración del nivel de conciencia y crisis comiciales. La evolución clínica es variable, algunos pacientes pueden entrar en coma y fallecer, otros pueden sobrevivir varios años con un cuadro de demencia o incluso algunos pueden recuperarse parcialmente23.
El diagnóstico es difícil por la gran variabilidad del cuadro clínico. De hecho, antiguamente el diagnóstico se basaba en estudios post mortem, pero, en la actualidad, el antecedente de alcoholismo, la clínica y, sobre todo, la neuroimagen cerebral, en concreto la RM, es esencial para confirmar el diagnóstico. Las lesiones típicas en RM son desmielinización, tumefacción y necrosis del cuerpo calloso, con extensión variable a la sustancia blanca subcortical24.
Dado que la etiología de la enfermedad es incierta, no se dispone de un tratamiento específico. Se recomienda el cese de la ingestión de alcohol y la suplementación vitamínica. Además, se ha comunicado algún caso de buena respuesta a dosis altas de corticoides25.
Demencia alcohólicaEl término demencia alcohólica se emplea para designar una forma de demencia atribuible a los efectos crónicos directos del alcohol sobre el encéfalo. Hay estudios que han demostrado que el consumo igual o superior a 140 g de alcohol al día durante un periodo prolongado de tiempo puede producir alteraciones cognitivas moderadas26.
Sin embargo, nunca se ha definido de manera precisa desde una perspectiva clínica ni patológica y, por ello, en los últimos años, el diagnóstico de demencia alcohólica ha desarrollado una controversia importante acerca de su entidad. Además, la interacción entre los déficits nutricionales, el consumo de otras sustancias, la comorbilidad psiquiátrica y los traumatismos craneoencefálicos repetidos en pacientes alcohólicos crónicos cuestionan la existencia de la demencia alcohólica pura. Algunos autores, por lo tanto, prefieren utilizar el término «daño cerebral relacionado con el alcohol» para reflejar la heterogeneidad de los trastornos cognitivos relacionados con el alcohol, tanto en la etiología como en la clínica.
Desde una perspectiva fisiopatológica, se cree que el daño o pérdida neuronal se relacionan con la neurotoxicidad glutamatérgica, estrés oxidativo y disrupción de la neurogénesis, desencadenados por el abuso crónico de alcohol27.
Estudios anatomopatológicos post mortem de casos diagnosticados de demencia alcohólica a menudo muestran hallazgos inespecíficos, como, por ejemplo, atrofia cerebral de predominio frontal, lesiones típicas del síndrome de Wernicke-Korsakoff, hidrocefalia comunicante, neuropatología de la enfermedad de Alzheimer o lesiones traumáticas de gravedad variable28.
Desde un punto de vista clínico, se caracteriza por un inicio insidioso con progresión escalonada de síntomas que se superponen con otras demencias neurodegenerativas. En fases iniciales, el estudio neuropsicológico suele poner de manifiesto un deterioro cognitivo de perfil frontosubcortical con enlentecimiento, déficit de atención, alteración de la memoria inmediata o a corto plazo, alteración visoespacial y alteración de las funciones ejecutivas de planificación y organización29.
Degeneración cerebelosa alcohólicaLa degeneración cerebelosa alcohólica es una complicación frecuente que afecta hasta a un 25% de los alcohólicos y es una de las causas más frecuentes de ataxia adquirida en el adulto30.
La patogénesis es compleja y no del todo conocida, aunque probablemente haya un mecanismo sinérgico, por lo que quizá esté implicado tanto el efecto tóxico del alcohol como las consecuencias del déficit de vitamina B1. Además, estudios recientes han demostrado la presencia de anticuerpos antitransglutaminasa tisular-2 en alcohólicos crónicos, lo cual plantea la posibilidad de una hipersensibilidad al gluten inducida por el alcohol31. Algunos autores han propuesto que una hipótesis que podría explicar esta hipersensiblidad al gluten sería que en pacientes alcohólicos se producen lesiones de la mucosa intestinal inducidas por alcohol, y esto podría aumentar la permeabilidad intestinal y promover la mayor exposición de nuevos antígenos (como los péptidos de gliadina), que se considerarían ajenos. Entonces, una alteración de la barrera hematoencefálica, en el contexto del consumo crónico de alcohol, por mecanismos aún poco conocidos, podría permitir el paso de dichos anticuerpos al cerebro y provocar una alteración y degeneración cerebelosa, de características similares a la ataxia cerebelosa por gluten32. De hecho, un estudio demostró que la prevalencia de anticuerpos antigliadina en pacientes con degeneración cerebelosa alcohólica es mayor que en la población general (44 vs. 12%)33.
Desde un punto de vista clínico, la degeneración cerebelosa se caracteriza por ataxia de tronco con marcha de base amplia e inestabilidad y grados variables de dismetría de extremidades inferiores. La dismetría en extremidades superiores, disartria o alteración oculomotora son menos frecuentes. En la mayor parte de los casos, el síndrome cerebeloso evoluciona durante un periodo de varias semanas o meses, tras lo cual permanece durante años.
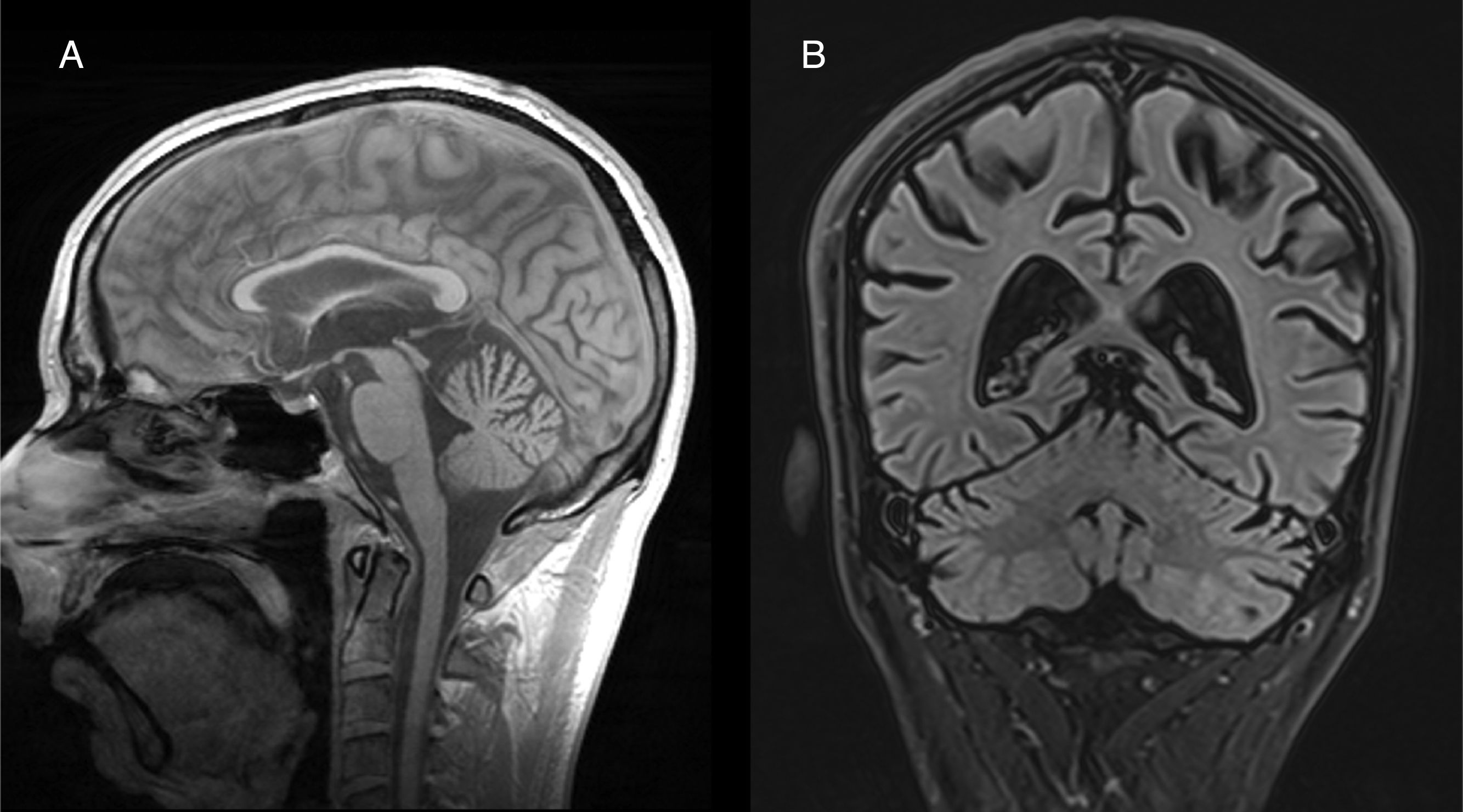
Tanto en estudios anatomopatológicos como de neuroimagen, hay una degeneración de todos los elementos neurocelulares de la corteza del cerebelo, pero, en particular, de células de Purkinje, en las superficies anterior y superior del vermis34. La atrofia del cerebelo se observa fácilmente mediante TC y RM cerebral (fig. 2). No hay un tratamiento específico, aunque se recomienda suplementación vitamínica y abstinencia alcohólica.
Afectación del sistema nervioso periféricoNeuropatía compresiva aguda y coronal (B) en FLAIR muestran atrofia cerebelosa de predominio vermiano.")
El consumo excesivo de alcohol se asocia clásicamente con el Saturday night palsy o «parálisis del sábado noche», debido a la compresión del nervio radial contra el canal de torsión del húmero durante unas horas. La causa suele ser el hecho de quedarse dormido con el brazo colgado o comprimido por el peso del cuerpo. Clínicamente se caracteriza por imposibilidad para realizar la flexión dorsal de la muñeca y extensión de los dedos. El estudio neurofisiológico mediante electromiografía es útil tanto para el diagnóstico como para el pronóstico, aunque la evolución habitual es hacia la recuperación en 3-6 meses35.
Polineuropatía crónica alcohólicaLa polineuropatía crónica es la complicación más frecuente en pacientes alcohólicos36. En cuanto a la etiología, de manera reciente hay mayor evidencia de que es un proceso multifactorial mediado principalmente por el efecto tóxico del alcohol modulado por otros factores como la predisposición genética, la deficiencia de tiamina, la malnutrición y otras enfermedades sistémicas37.
Es una polineuropatía predominantemente axonal, sensitivo-motora, distal y simétrica. El inicio de los síntomas es insidioso, de predominio sensitivo y simétrico, en forma de disestesias, sensación de quemazón y dolor urente en las plantas de los pies, molestias que más tarde se difunden en las pantorrillas con calambres y también en las manos. La sintomatología motora suele ser más tardía, y se caracteriza por debilidad y atrofia muscular, sobre todo de la musculatura distal de las extremidades superiores o inferiores. Además, también son características las alteraciones vegetativas cutáneas y vasomotoras (piel sudorosa, atrófica, brillante y con escaso vello), así como la disautonomía asociada.
El tratamiento consiste en mantener abstinencia alcohólica, una dieta equilibrada con suplementos vitamínico y rehabilitación. Sin embargo, la recuperación es lenta y a menudo incompleta. Si existe dolor de características neuropáticas pueden utilizarse fármacos como la gabapentina o amitriptilina.
Neuropatía por disulfiramEl disulfiram, fármaco empleado para mejorar la abstinencia alcohólica, se ha asociado, en raras ocasiones, a una neuropatía periférica. El mecanismo fisiopatológico por el cual se produce es desconocido. Es una polineuropatía axonal, sensitivo-motora y dosis dependiente, que empieza unas semanas o meses tras el inicio de la toma del fármaco. Desde el punto de vista clínico se caracteriza por parestesias distales en distribución de guante y calcetín, junto con debilidad muscular de predominio distal y arreflexia distal. El pronóstico se relaciona con la gravedad y grado de pérdida axonal, aunque generalmente es reversible tras el cese del fármaco38.
Afectación muscularMiopatía alcohólica aguda o crónicaEl alcohol puede tener efectos nocivos sobre el músculo esquelético por la alteración de los canales de calcio o de la integridad de la membrana de la fibra muscular o sarcolema. La miopatía alcohólica puede cursar clínicamente en forma de un cuadro agudo con mialgias, rabdomiólisis e elevación de la creatincinasa (CK) y puede ser grave y llegar a condicionar fallo renal agudo y mioglobinuria. En las formas crónicas aparece una atrofia muscular generalmente de distribución proximal que frecuentemente coexiste con alteraciones neuroperiféricas como polineuropatía crónica alcohólica. El diagnóstico se realiza tanto con estudio neurofisiológico mediante el electromiograma, como histopatológico con la biopsia muscular. En algunos casos la miopatía esquelética se acompaña de miocardiopatía dilatada. El tratamiento se basa en la abstención alcohólica, fisioterapia y nutrición adecuadas, y el pronóstico es reservado, ya que algunos pacientes experimentan mejoría de la debilidad clínica, pero otros no acaban de recuperar ni la masa ni la fuerza muscular39.
La encefalopatía hepáticaLa EH es una complicación grave y frecuente de la cirrosis hepática y un indicador de mal pronóstico en estos pacientes. Es un complejo síndrome de alteraciones neuropsiquiátricas que se produce en pacientes con insuficiencia hepática avanzada o derivación sanguínea portosistémica40.
La clínica es variable y fluctuante, y abarca desde síntomas como temblor y disartria hasta coma hepático. Incluye a) alteración del nivel de conciencia, que puede progresar desde un estado confusional leve hasta un estado de coma; b) síntomas neuropsiquiátricos como cambios de comportamiento, lentitud mental, inversión del ciclo sueño-vigilia o agitación psicomotriz y c) signos neuromusculares entre los que destaca el temblor aleteante o flapping. Existe una clasificación clínica clásica de West Haven que estadifica la EH en 4 grados de menor o mayor gravedad (tabla 3). Dado que las manifestaciones clínicas de la EH no son específicas y pueden observarse en otras enfermedades o trastornos metabólicos, el diagnóstico se realiza tras la exclusión razonable de otras causas potenciales mediante la realización de exploraciones complementarias.
Grados de encefalopatía hepática de West Haven
| Grados de encefalopatía hepática |
|---|
| I Euforia, ansiedad, disminución capacidad de atención, inversión del ritmo sueño-vigilia. Flapping esporádico |
| II Letargia, apatía, desorientación temporoespacial, trastorno de la conducta. Flapping evidente |
| III Somnolencia profunda, estupor, estado confusional, conductas inapropiadas, desorientación importante. Flapping en ocasiones inexplorable por falta de colaboración |
| IV Coma |
En pacientes con alcoholismo crónico, la EW plantea un reto diagnóstico, ya que el diagnóstico diferencial es extenso.
Desde un punto de vista clínico, cabe destacar que el diagnóstico de EW puede ser difícil ya que, como se ha explicado con anterioridad, a menudo no está presente la tríada clásica. La presencia de alteraciones de la motilidad ocular, nistagmo y ataxia debe hacer sospechar la EW, mientras que la presencia de flapping o piramidalismo, la EH. En cuanto a la abstinencia alcohólica, algunos hallazgos clínicos que podrían señalar el diagnóstico son la presencia de ansiedad, taquicardia, alucinaciones visuales y temblor postural. La enfermedad de Machiafava-Bignami en general no plantea dificultad en el diagnóstico diferencial con la EH, ya que clínicamente la primera se caracteriza por la presencia de demencia y espasticidad.
La neuroimagen cerebral mediante tomografía computarizada (TC) o RM permite descartar alteraciones estructurales como lesiones ocupantes de espacio, hematomas subdurales o ictus isquémicos/hemorrágicos, que pueden sospecharse mediante la objetivación de focalidad neurológica en la exploración. Además, también permite descartar encefalitis víricas o autoinmunes que clínicamente pueden ser superponibles a la EH. En la EH destaca un hallazgo típico en RM que consiste en una señal hiperintensa de los ganglios de la base en secuencias potenciadas en T1, sobre todo en el globo pálido, que se relaciona con depósitos de manganeso por la presencia de shunts portosistémicos, y que podrían justificar la existencia de signos parkinsonianos en estos pacientes41.
Por otro lado, el registro electroencefalográfico, prueba neurofisiológica que traduce la actividad eléctrica cerebral, permite descartar un estatus no convulsivo o hallazgos típicos de un estado poscrítico. No obstante, en la EH se producen alteraciones de la actividad cerebral que traducen cambios en el electroencefalograma en forma de ondas lentas con incremento de su amplitud y ondas trifásicas, hallazgos que también pueden encontrarse en otros comas de origen metabólico42.
Por último, el estudio del líquido cefalorraquídeo permite descartar meningitis o meningoencefalitis bacteriana o vírica, que debería sospecharse en caso de fiebre y signos meníngeos.
ConclusionesEl consumo crónico de alcohol puede producir numerosas manifestaciones neurológicas. Las más frecuentes son la polineuropatía, la degeneración cerebelosa y la demencia, y las más graves la EW, el síndrome de Korsakoff y la enfermedad de Machiafava-Bignami. Todas ellas se relacionan con una significativa morbimortalidad, por lo que su correcto diagnóstico y tratamiento es esencial para evitar complicaciones irreversibles. Los mecanismos por los cuales se producen incluyen déficits vitamínicos, efectos tóxicos directos del alcohol, alteraciones inmunes y mecanismos desconocidos. Además, la EH puede plantear dificultades en el diagnóstico diferencial con alteraciones neurológicas asociadas al alcoholismo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Mostramos nuestro agradecimiento al Instituto de Diagnóstico de la Imagen (IDI), Unidad de Resonancia Magnética, Hospital Germans Trias i Pujol (Badalona, Barcelona, España), en especial a Fidel Núñez.

