




Thalassemias are the most frequent monogenic disorder around the world. α-Thalassemias are due to a deficiency of synthesis in the alpha-globin chain of the hemoglobin (Hb). Hb Groene Hart is a hyperunstable variant. In this work, we have studied 24 cases affected by Hb Groene Hart, one of them associated with Hb J-Paris-I.
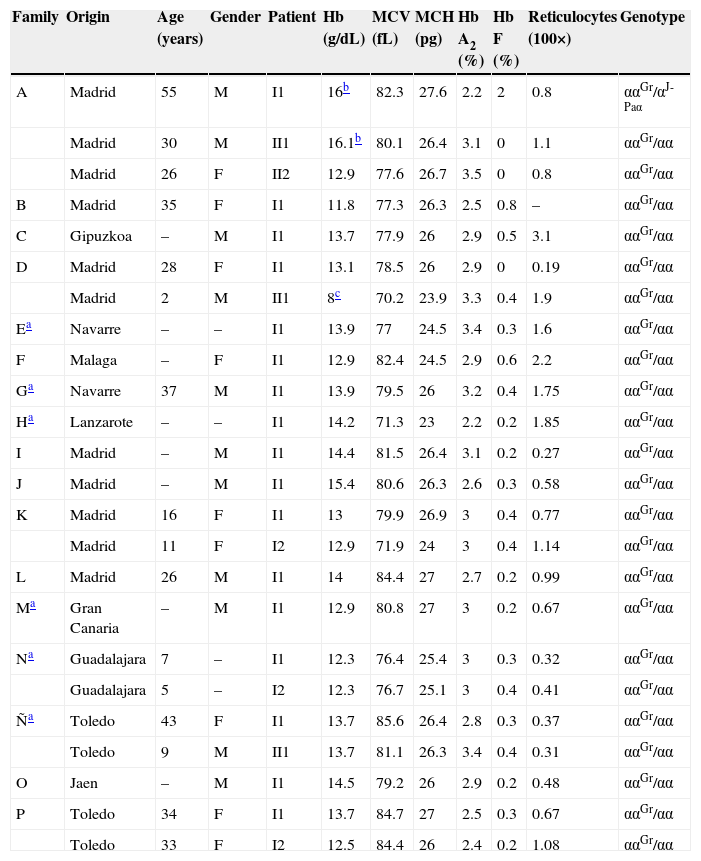
Patients and methodsTwenty-four patients from 17 unrelated families were included in this study. The characterisation was done by sequencing.
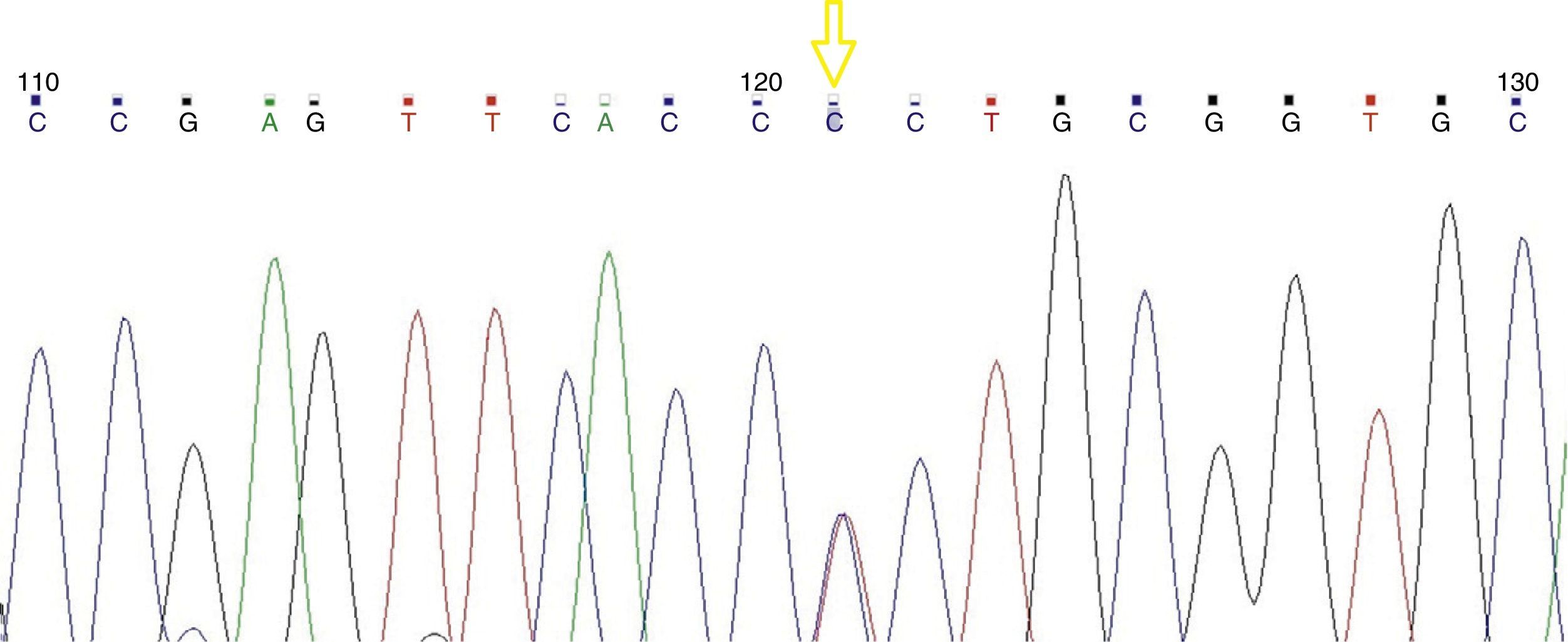
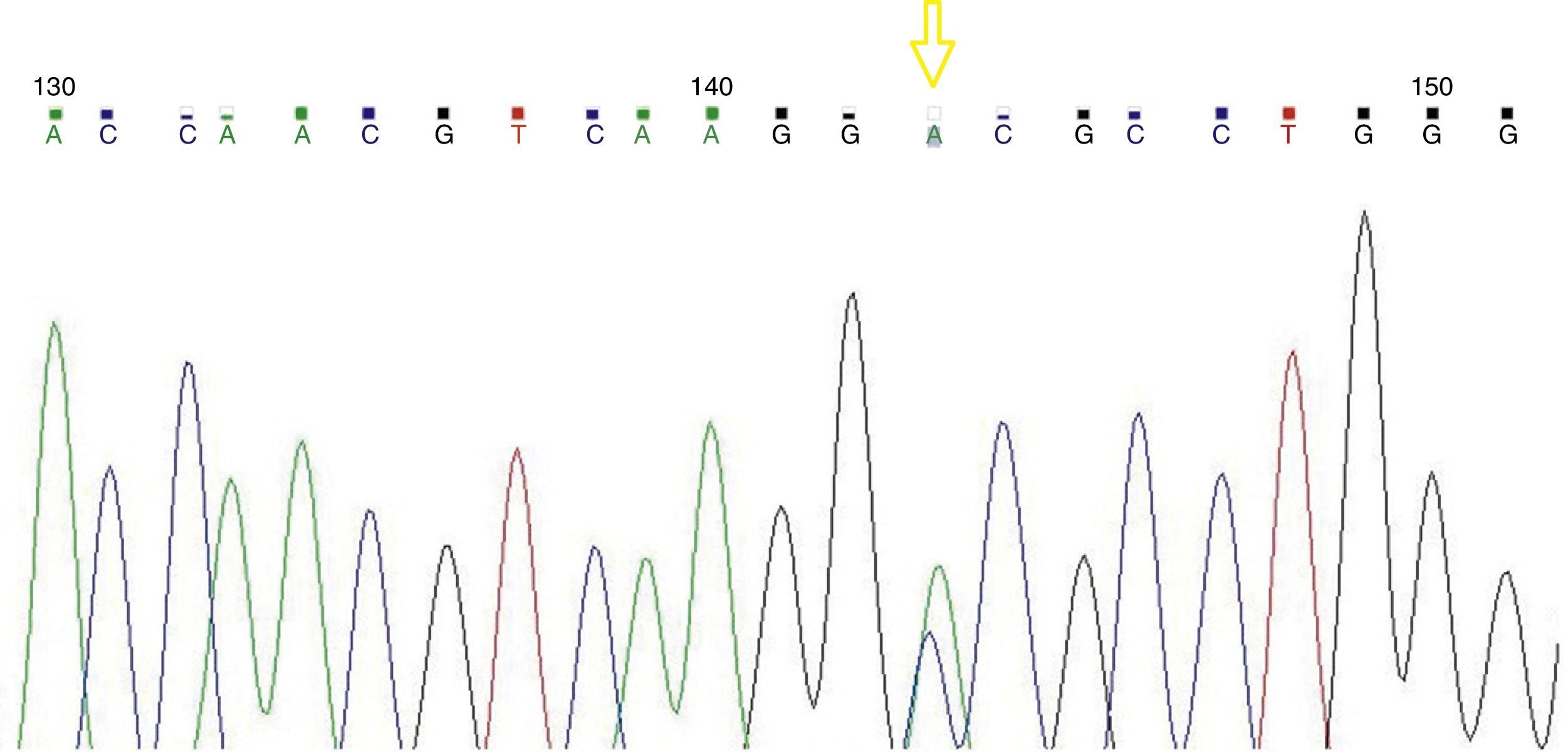
Resultsα1 gene sequencing showed the mutation CCT→TCT (Pro→Ser) at codon 119 (Hb Groene Hart) in all patients. In one case, there was an association with Hb J-Paris-I.
ConclusionsIn the Hb Groene Hart, the residue 119 of alpha-globin chain is affected. This amino acid has a key role in preserving the stability of alpha-globin chain. It is also remarkable the presence of this variant in both the immigrant and native population. Thus, the identification of Hb Groene Hart carriers should be considered in the screening of α-thalassemia in Spain, as it is done in Northern Africa.
Las talasemias son las enfermedades monogénicas más frecuentes a nivel mundial. Representan un grave problema sanitario en las regiones de mayor incidencia. Las α-talasemias se deben a un déficit de síntesis de las cadenas α de la hemoglobina (Hb). La Hb Groene Hart es una variante hiperinestable. En este trabajo se presentan 24 casos pertenecientes a 17 familias afectadas por la Hb Groene Hart, uno de ellos asociado con Hb J-París-I.
Pacientes y métodoVeinticuatro pacientes de 17 familias no relacionadas fueron incluidos en este estudio. La caracterización se realizó mediante secuenciación.
ResultadosLa secuenciación del gen α1 mostró la mutación CCT→TCT (Pro→Ser) en el codón 119 (Hb Groene Hart). En un paciente se asoció con la Hb J-París-I.
ConclusionesEn la Hb Groene Hart se encuentra afectado un residuo clave para preservar la estabilidad de las cadenas α de globina. La presencia de esta variante es elevada en población española e inmigrante. La aparición de formas graves de la enfermedad podría ser evitada incorporando esta mutación al cribado de las mutaciones α-talasemia no deleción.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
