




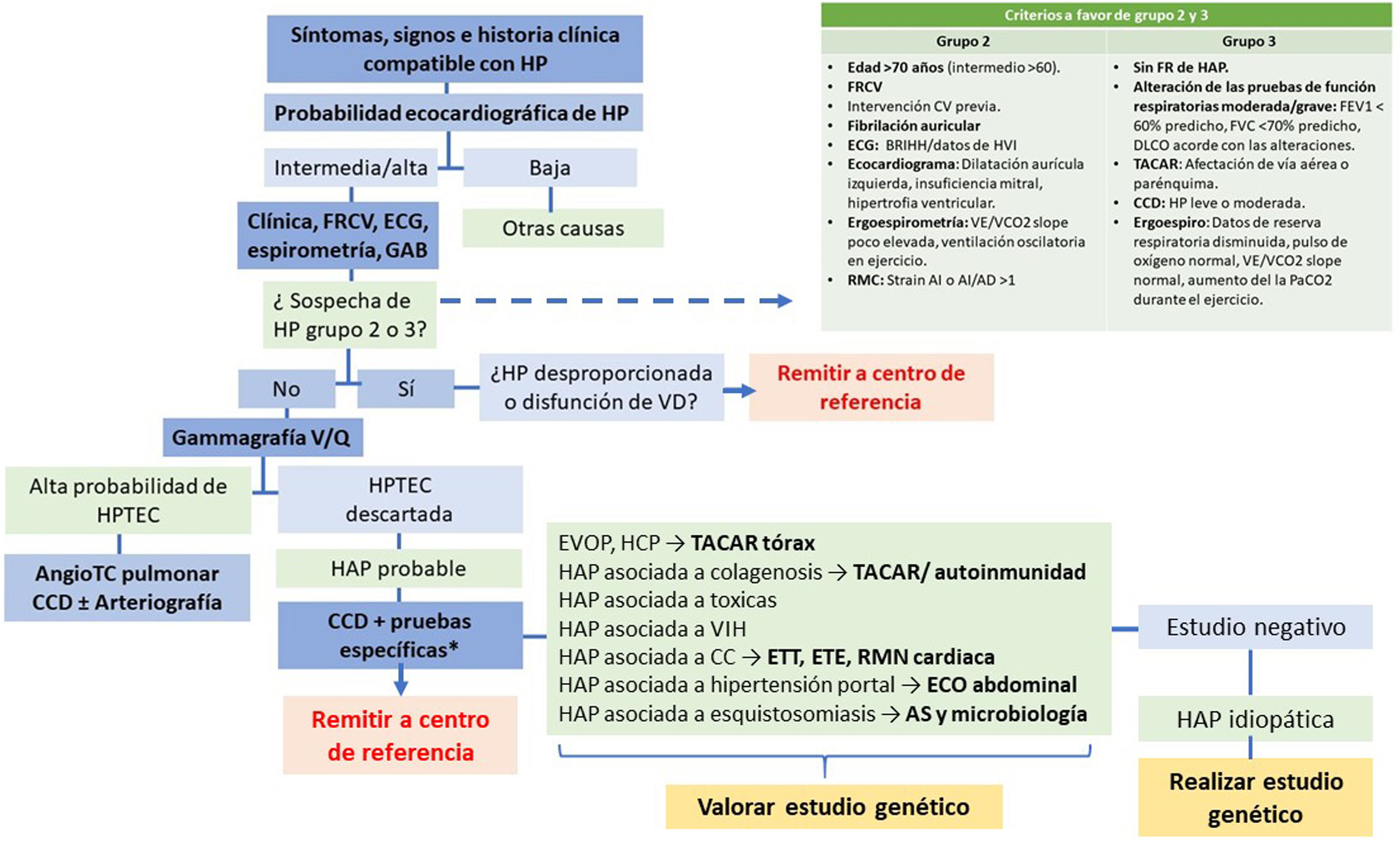
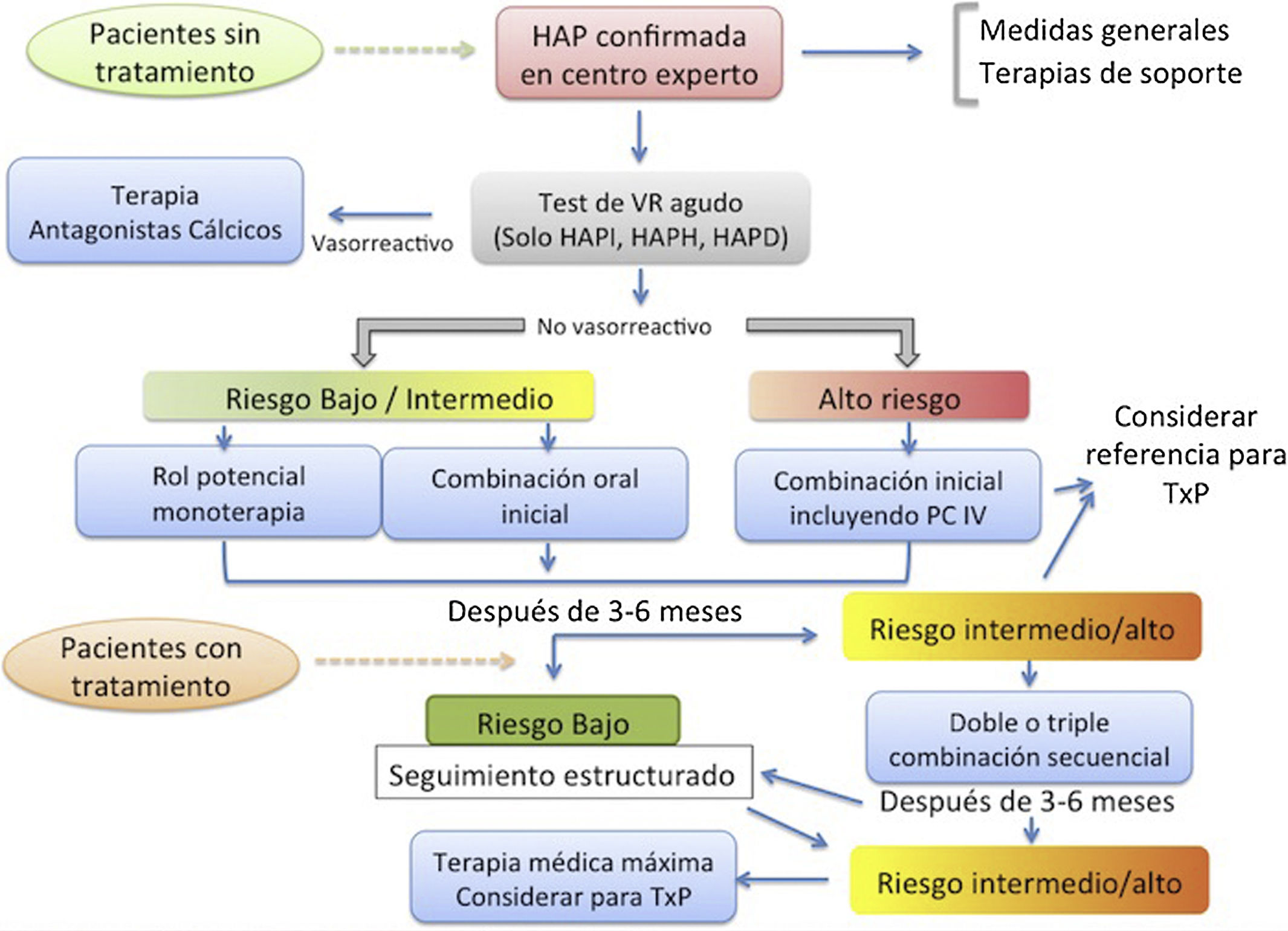
La hipertensión arterial pulmonar (HAP) es una enfermedad rara caracterizada por un remodelado adverso del árbol arterial que conlleva un aumento de las resistencias vasculares con el subsecuente incremento de la poscarga del ventrículo derecho y el desarrollo, finalmente, de insuficiencia cardiaca. El curso de la enfermedad conlleva un mal pronóstico, al que se asocia el retraso en el diagnóstico e inicio de tratamiento motivado por la clínica inespecífica y el desconocimiento de la entidad. Las recomendaciones más recientes se centran en optimizar el diagnóstico precoz diferencial frente a otras causas de hipertensión pulmonar, con el fin de iniciar un tratamiento adecuado utilizando como referencia la estimación de riesgo de mortalidad del paciente. En los últimos años, con la mejora en el proceso diagnóstico, la aparición de nuevos tratamientos específicos y la creación de unidades de referencia especializadas en esta enfermedad, se ha conseguido mejorar el pronóstico y la calidad de vida de los pacientes con HAP.
Pulmonary arterial hypertension (PAH) is a rare disease characterized by adverse remodeling of the arterial tree leading to increased vascular resistance with subsequent increase in right ventricular afterload and eventual development of heart failure. The nonspecific clinical manifestations and lack of knowledge of pathology lead to a poor prognosis associated with delay in diagnosis and initiation of treatment. The most recent recommendations focus on optimizing the early differential diagnosis with other causes of pulmonary hypertension to initiate appropriate treatment based on the mortality risk estimation. In the last years, with the improvement in the diagnostic process, the emergence of new specific treatments, and the creation of specialized referral units for this pathology, the prognosis, and quality of life of patients with PAH have improved significantly.

