



Congenital long QT syndrome (cLQTS) is a channelopathy characterized by ventricular repolarization disturbances. The clinical presentation varies from an asymptomatic patient to a patient with recurrent syncopes, seizures, and even sudden death. This article aims to contribute to medical knowledge of this relatively new disease in newborns, so that early diagnoses with timely treatments can be made. Genotypically, there are 13 types of cLQTS, which are classified by phenotype into: Romano-Ward, Jervell-Lange-Nielsen, Andersen-Tawil and Timothy. We present the following case of a newborn who presented clinical sustained bradycardia on her first day of life, who was admitted to the Neonatal Intensive Care, where her assessment and imaging studies guided us to a cLQTS diagnosis. The patient recovered satisfactorily, and was discharged with outpatient follow-up without complications.
All abnormal QT patients should be assessed with an integral clinical report, Holter analyzer, and a stress test. Ideally, a genetic screening, which can have a great impact on the treatment, should be done. Although it is relatively uncommon, recognition of this entity is important because it can prevent death. We present a case of cLQTS diagnosis, with approach and treatment.
Neonatal arrhythmias represent a relatively common problem encountered by pediatricians. However, because most of them turn out to be benign, it is vital to master the diagnostic approach which allows the identification of patients who may be at risk of death.1 cLQTS is a rare genetic entity from the denominated channelopathies, characterized by mutations in ion channels or their regulatory proteins. It may lead to sudden death because of ventricular repolarization alterations, which is seen in an electrocardiogram as a long QT.2
This alteration is the main cause of sudden death in children; this is secondary to a malignant ventricular tachycardia (torsade de pointes), and can be often be provoked by a series of conditions, including sudden noise, exposure to cold environments, REM sleep, apnea, leading a chemoreceptor reflex and excitement. It is also important to obtain a detailed family history and pay special attention to sudden deaths, ventricular ectopic beats and seizures, since these may be present in up to 35% of the seizures induced by long QT and could be mistaken for other causes of seizures.3 Long QT acts as an arrhythmogenic substrate, in other words, it requires a trigger for the development of life-threatening arrhythmias, like catecholamines or medications which delay QT intervals even more. Examples of these medications are listed in Table 1. A genetic study in all patients with said electrocardiographic alteration must be performed.4 We present a clinical case of a newborn with an abnormal QT prolongation.
Clinical caseNewborn without any important hereditary-family history, full term (38.6 weeks), delivered via C-section due to pelvic presentation. The baby only required the initial steps of reanimation, with an Apgar score of 8/9. A newborn routine was performed registering a weight of 3180g, 49cm in length and a head circumference of 34cm. However, a few hours after she was born, we detected an 80 beat-per-minute bradycardia; therefore, we decided to admit her to the Neonatal Intensive Care Unit (NICU). During physical examination we recorded a heart rate of 74 beats-per-minute, a respiratory rate of 54 breaths per minute, blood pressure of 70/60mmHg and SaO2 of 96%. The following dysmorphias were found: Head with wide forehead, wide nasal bridge, low-implanted ears, thin upper lip, enlarged philtrum and micrognathia; extremities with clinodactyly in the 5th bilateral finger and bilateral transverse crease; skin with superficial scaling on the face and scalp, in addition to eczematous lesions in the cheeks. The rest of the physical examination was normal.
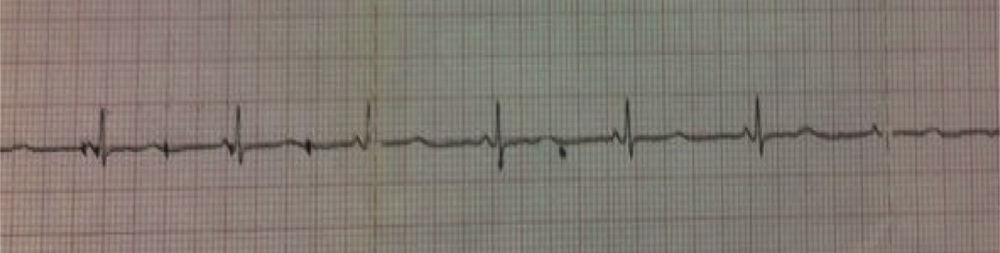
The admission tests, such as biochemical profile, serum electrolytes and complete blood count showed no alterations, as was the case for thoracic X-rays too. The ECG showed sinus bradycardia and we measured the QT using the Bazzet formula finding a long QT in 0.46s (Figs. 1 and 2).


The patient was referred to Pediatric Cardiology where they performed an echocardiogram, reporting a permeable foramen ovale of 2mm without hemodynamic repercussion and performed Holter monitoring presenting non-sustained sinus bradycardia with a minimum HR of 58 (bpm) and alternating T waves. With this data, we integrated a sinus bradycardia with long QT diagnosis and we performed consults with the Genetics Service who suspected Andersen–Tawil syndrome. The patient received treatment with beta-blockers (propranolol 3mg/kg/day orally divided into three doses) evolving to be hemodynamically stable without presenting bradycardias. We considered a discharge after 13 days of life outside the womb.
DiscussionChannelopathies are the main cause of congenital arrhythmias. They are clinical entities in which there is a mutation of the genes that control the formation of transmembrane protein responsible for regulating the amount of ions that go through the cell.5 cLQTS is the first channelopathy described and it is currently considered the most studied. It is characterized by a ventricular affectation of the repolarization phase and can cause sudden death by polymorph ventricular tachycardia or Torsade de Pointes.6 It was first described in 1957 by Anton Jervell and Fred Lange Nielsen, who linked a syndrome characterized by congenital deafness, syncope and sudden deaths with abnormal prolongation of the QT interval. Later, in 1964, Romano and Ward observed similar clinical characteristics but without deafness.
Clinical presentation varies from the asymptomatic patient, to the patient with recurrent syncopes, seizure crises and even sudden death. Its incidence is 1/5000 cases.7,8 To this day, over 500 mutations have been identified, distributed in 10 genes. Genotypically, there are 13 types of cLQTS, and by phenotype, and they can be classified into four different ways: Romano-Ward, Jervell-Lange-Nielsen, Andersen-Tawil and Timothy.9
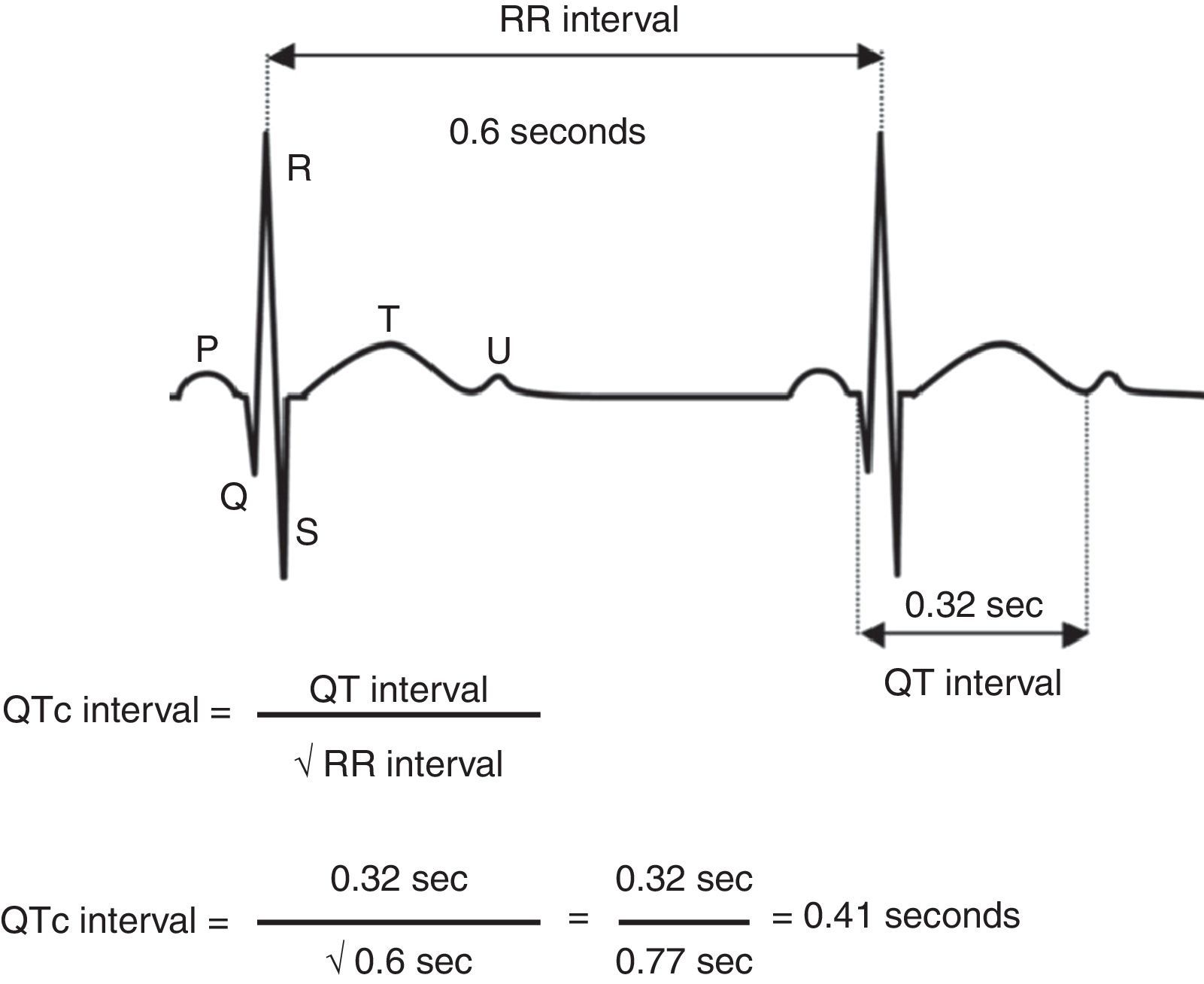
Corrected QT interval measurement was obtained using Bazett's formula, dividing the QT interval by the square root of the RR interval (cQT=QT/√RR) ideally in the V5 or DII derivations. In diagnostic orientation Schwartz's score takes into account electrocardiographic findings, family history and pathological background like syncopes.10
In this clinical case we phenotypically describe minor facial dysmorphias such as: A wide forehead, low-implanted ears, a thin upper lip and micrognathia, in addition to clinodactyly in the 5th bilateral finger, which fits better in a description of cLQTS 7 or Andersen Tawil. This syndrome currently is considered to be a different entity by many authors. It is an autosomal dominant alteration, predominant in females and phenotypically characterized by clinodactyly, hypertelorism, wide forehead and low ear implantation. Periodic paralysis, abnormal skeletal development and ventricular arrhythmias may occur. Patients tend to be asymptomatic, or minimally symptomatic despite having frequent extrasystoles or ventricular tachycardia, typically bidirectional or polymorphous.11
The beta-blockers, which have been used for over 30 years, reduce global mortality from 73% to 6%. Currently, all patients diagnosed with cLQTS, with or without clinical symptoms, must receive beta-blocker treatment, and in addition the patients are advised to avoid rigorous physical activity and administration of medications which prolong repolarization (Table 1). In patients with high sudden-death risk an implantable automatic defibrillator (IAD) must be prescribed. Within this group we included patients who were symptomatic despite beta-blocker treatment, patients with auriculo-ventricular blocks, those with documented ventricular tachycardia in torsade de pointes, and those patients resuscitated from sudden death. It should also be prescribed in patients with an excessively long cQT (≥0.5s) and when there is a family history of sudden deaths.12
Aside from the diagnostic suspicion to which phenotypic features may lead us, all abnormal corrected QT patients must be approached with a careful clinical history, and Holter monitoring. We recommend a follow-up by cardiologists and geneticists, and ideally, a genetic screening, which can have an impact on treatment according to the mutations found.
ConclusionThe importance of this case is to create awareness in the medical community of these new pathologies which represent some of the most important causes of neonatal arrhythmias; however, this information, in addition to clinical history, would be of assistance to keep in mind in these entities, thus having early diagnosis and treatment, avoiding death. Moreover, we stress the importance of complementing it with geneticists in order to make better decisions with the families. Even though cLQTS is relatively rare, to recognize this entity is of great importance, because early intervention and treatment can avoid the patient's sudden death.
Conflict of interestThe authors have no conflicts of interest to declare.