


Congenital fiber-type disproportion myopathy causes impaired muscle maturation or development. It is characterized by moderate to severe hypotonia and generalized muscle weakness at birth or during the first year of life, especially in the lower extremities. It is inherited as an autosomal recessive, dominant and X-linked. It is diagnosed by clinical data confirmation, generalized hypotonia and a muscle biopsy in which muscle fibers type I are smaller in caliber, 12% smaller than those of type II and type I fibers are more common than type II. Treatment is multidisciplinary.
The following describes the case of a patient who was born in the “Dr. José Eleuterio González” University Hospital in Monterrey, N.L, who presented clinical and muscle biopsy compatible with this myopathy.
Within congenital myopathies classifications, there are myopathies with alterations in muscle maturation and/or development. One of these myopathies is congenital fiber-type disproportion, which is characterized by generalized muscle weakness at birth or during the baby's first year, mainly in the extremities.1,2
It was first described by Brooke and Engel1,3 in a study of the morphology of children's biopsies. It is a rare entity, which occurs in 1 in every 50,000 live births.4 All body musculature is affected. However, the muscles in the lower extremities are usually more altered than the upper extremities; thus the electromyography reports a myopathic process, although in others there is a neurogenic component.11,13 Some cases with a longer evolution time show mild sclerosis5 and there have been reports of important respiratory alterations, severe brain damage and heart diseases.1,7,8 In a few cases an association of skeletal and articular alterations has been observed.9,10
Clinical caseThe mother of the baby boy was a 25-year-old woman, previously healthy, secondary school completed, without drug addictions, living together with her partner. The father was a 30-year-old man, healthy, without any relevant history. The newborn had two other brothers, a 4-year-old and a 7-year-old, both healthy.
The product of a fourth pregnancy, the mother had had two C-sections performed and suffered a miscarriage when she was 9 weeks pregnant. She completed a normal pregnancy, with folic acid, iron intake, as well as supplementary multivitamins for the first month of pregnancy, prenatal control with 8 visits to the University Hospital without presenting abnormalities.
Born by elective C-section due to the history of two previous C-sections at 38.4 weeks of gestation, with an Apgar score of 2/6 because it was found to be acrocyanotic, flaccid, no respiratory effort, and with a heart rate of 80bpm. He is given 2 positive pressure ventilation cycles, improving hear rate, but not respiratory effort, and was therefore intubated and left under mechanical ventilation and moved to the Neonatal Intensive Care Unit.
During physical examination: Weight: 3070g, located in the 50th percentile, size: 50cm, in the 50th percentile, cephalic perimeter: (CP) 36cm, in the 50th percentile, myopathic facies with an elongated face, symmetrical eyes and a bilateral ptosis, presenting red reflex, permeable nostrils, proper ear implantation, an ogival palate and a tented upper lip. Neck: retrognathial, short, round. Thorax: no respiratory effort, without deformities, normal lung fields, holosystolic murmur grade 3–4 detected. The abdomen was soft, palpable without pain, and free of visceromegaly, an umbilical cord, 2 arteries and a vein. Male genitalia: Tanner 1, right cryptorchidie, with the right testicle in the inguinal canal and the left testicle in the scrotal bag. The extremities: generalized hypotonia, no response to painful stimulus, absence of muscle tone, deep tendon reflexes absent, rigid right clubfoot.
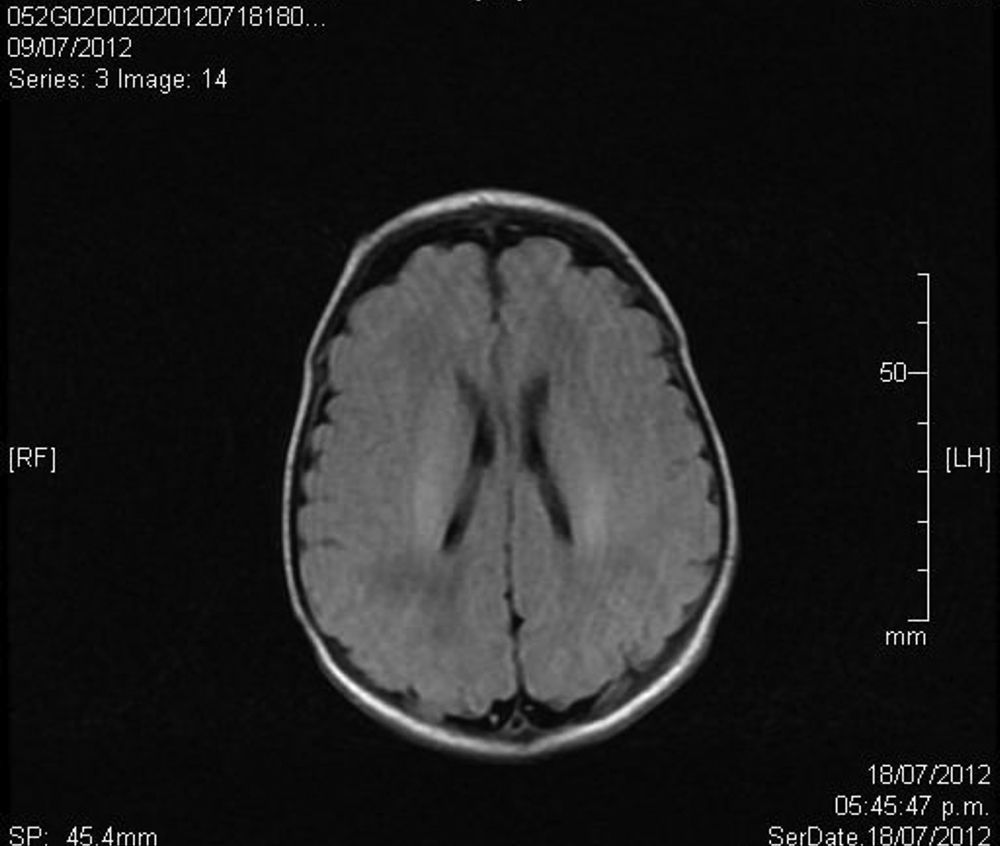
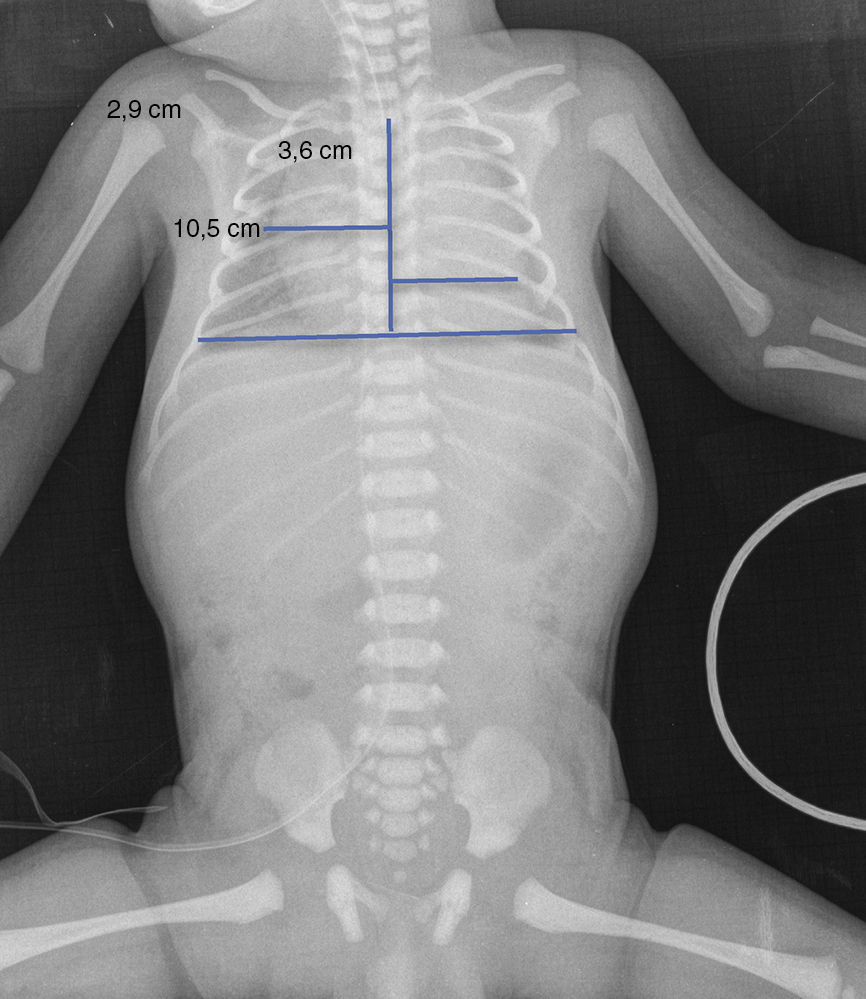
Upon admission, gasometry and asphyctic profile on admission and at 24h (due to perinatal asphyxia suspicion) were normal and a transfontanellar ultrasound (TFUS) and brain MRI were performed and were also normal (Fig. 1). No infection-compatible alterations were found, as well as the metabolic panel (with electrolytes) and the metabolic screening were also normal. An electrocardiogram was performed due to the presence of ICT in 0.62 (Fig. 2) and due to the holosystolic murmur grade II/VI detecting an interventricular communication of 2mm, mild tricuspid insufficiency, a persistence of a patent ductus arteriosus of 1.4mm and an oval foramen without hemodynamic repercussions.


We referred the patient to the Genetics Department who evaluated the patient's genealogical tree, without finding any history of neuromuscular diseases in any of the first-degree relatives or extended family, thus classifying this as an isolated case. Furthermore, they requested a genetic mutations study; however, the family did not have the economic resources to have it done.
Due to the newborn's hypotonic diagnosis, a muscle biopsy, lab tests and normal imagining of the right quadriceps were performed, confirming the congenital fiber-type disproportion diagnosis.
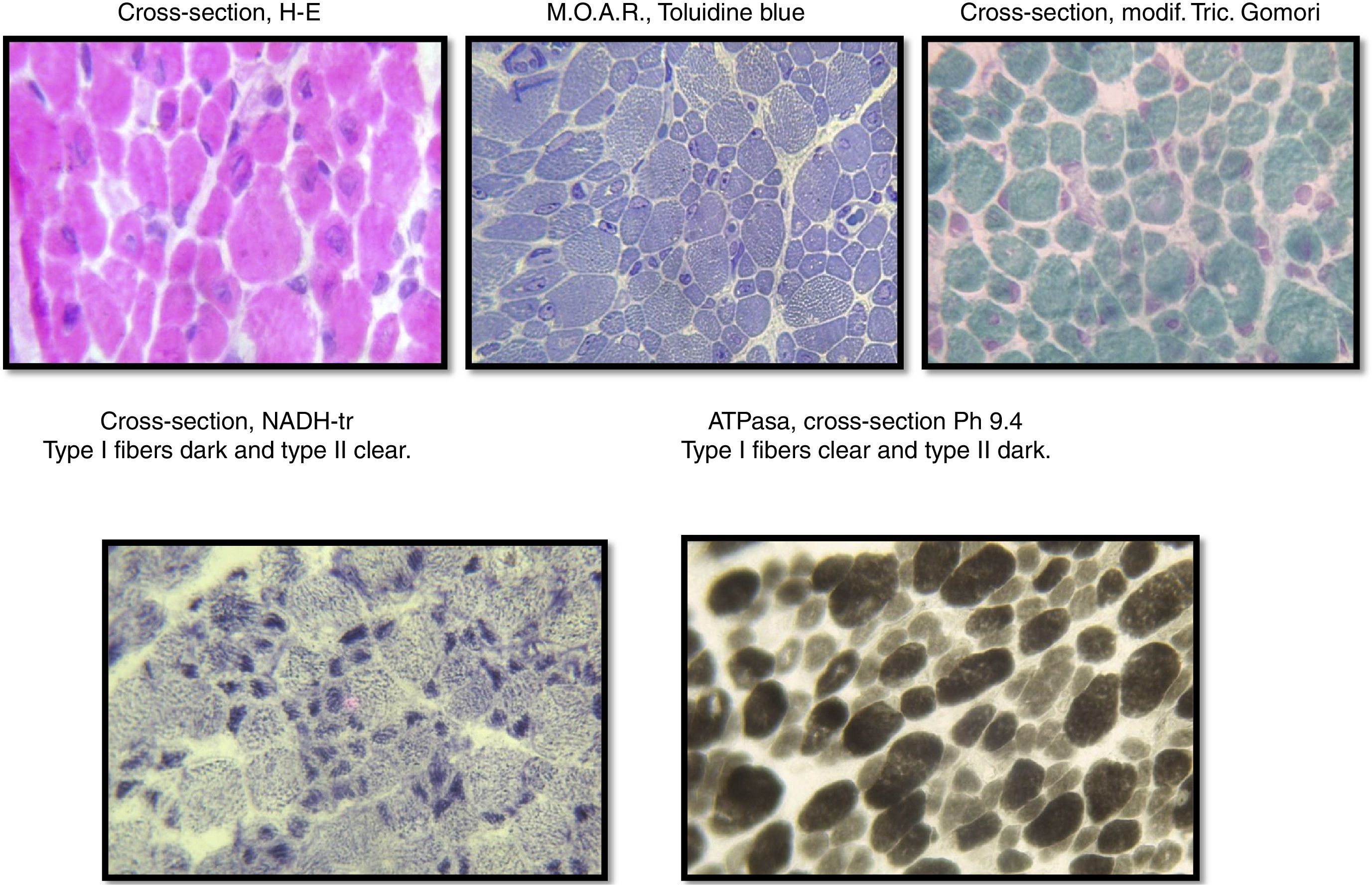
The sample was divided into two parts; the first part was cut by freezing it and processed following standard and special techniques for its morphological, histochemical and enzymatic analyses, applying H&E stains, a modified Gomori trichrome and ATPase reactions at pH 4.3, 4.6 and 9.4, Nicotinamide dinucleotide and adenine-tetrazolium reductase (NADH-tr). In addition, we conducted a morphological study using light microscopy and high-resolution optics microscopy. A morphometric analysis was conducted on type-I and type-II fibers. Findings showed the following: it was determined that, regarding type-I fibers, 20% of them were within the normal interval (10–15.4μm) with an average of 6.9μm (normal of 12.7μm) and a variable coefficient of 449 (normal up to 250) due to the 80% of atrophic fibers, with a very elevated atrophy coefficient of 1,350 (normal up to 150). For type-II fibers, the average was 15.4μm (normal 10.9μm) with just 38% of fibers within the normal interval (7.7–14.1μm), the variable coefficient does not exceed the limit value {241 (normal up to 250)} despite the high percentage of hypertrophic fibers (62%) but the hypertrophy coefficient is very elevated {600 (normal up to 400)} (Fig. 3).
Discussion
Congenital fiber-type disproportion myopathy, as displayed in our patient in this case, is determined by the unevenness in the size of both types of muscle fibers, where type-I fibers are much smaller, and type-II fibers are too large.1,2 According to the study by Brooke and Engel of the morphology of biopsies, the entity is based histopathologically in a threshold of 12% of unevenness in fiber sizes to define the group.13
Our patient's case is relevant because congenital fiber-type disproportion myopathy is a rare entity, which according to medical literature occurs only in 1:50,000 live births.3,13 The most common clinical manifestations in 50–75% of cases are moderate to severe hypotonia (floppy babies), general muscle weakness at birth or during the first year, especially in the lower extremities, and absence of deep tendon reflexes, which the patient displayed in our case, normal or slightly increased creatine kinase (CK), normal intelligence, as well as normal electromyography or myopathic pattern.3,6 Common clinical manifestations, which are present in 10–50% of cases are: myopathic facies with an elongated face, an ogival palate, a tented upper lip and light to severe respiratory problems, all of which were found in our patient, difficulty feeding, ophthalmoplegia, contractures, scoliosis, lordosis and laxity.11,12 The less frequent manifestations, present in less than 10% of cases are: cardiac involvement, cognitive impairment and cryptorchidism, which was present in our clinical case.3,6
It is a genetically heterogeneous disease, which in inherited in an autosomal recessive manner, dominant and with isolated cases.1,2,9,12 To date, mutations in six genes have been identified: ACTA1 (6%), MYH7 (unknown), RYR1 (10–20%), SEPN1 (rare), TPM2 (rare) and TPM3 (20–25%).16
During the muscular biopsy, we found type I muscular fibers of a smaller caliber (12% smaller) than the type IIs, which were found to be hypertrophic. And in the same way we found the type I or atrophic fibers, we also found a greater number of type II or hypertrophic14,15 fibers, which were found in the immunohistological report of the muscular biopsy performed in the case of our patient.
The best threshold for the level of disproportion of muscle fibers necessary to make a diagnosis is still under discussion, but there is a general consensus that it should be equal to or greater than 12%. The prenatal diagnosis is made by analyzing DNA extracted from fetal cells with a chorionic villus sampling at 10 or 12 weeks of gestation and an amniocentesis at 15 to 18 weeks of gestation, where the mutation in some of the 3 genes causing the disease can be found.3,13
The treatment is multidisciplinary, with physical rehabilitation and treatment of complications such as respiratory difficulties, joint alterations, weakness and swallowing difficulties.3,13
The prognosis depends on the degree of hypotonia as well as respiratory and cardiac involvement. Ophthalmoplegia, ptosis, a bulbous or weak face, severe weakness in the extremities and respiratory weakness have been described as factors for a negative prognosis.3,11,13
The presentation of this clinical case is important, as it deals with an infrequent entity, which represents a diagnostic and therapeutic challenge in our line of work. It is important to get to know the clinical manifestations, how to arrive at a diagnosis, and understand the complications which may arise in these patients, to provide adequate treatment, prevent complications, and offer a better quality of life. Additionally, the presentation of this case opens the doors to more diagnostic possibilities which we should look forward to cope with a newborn with generalized hypotonia.
Conflict of interestThe authors have no conflicts of interest to declare.