


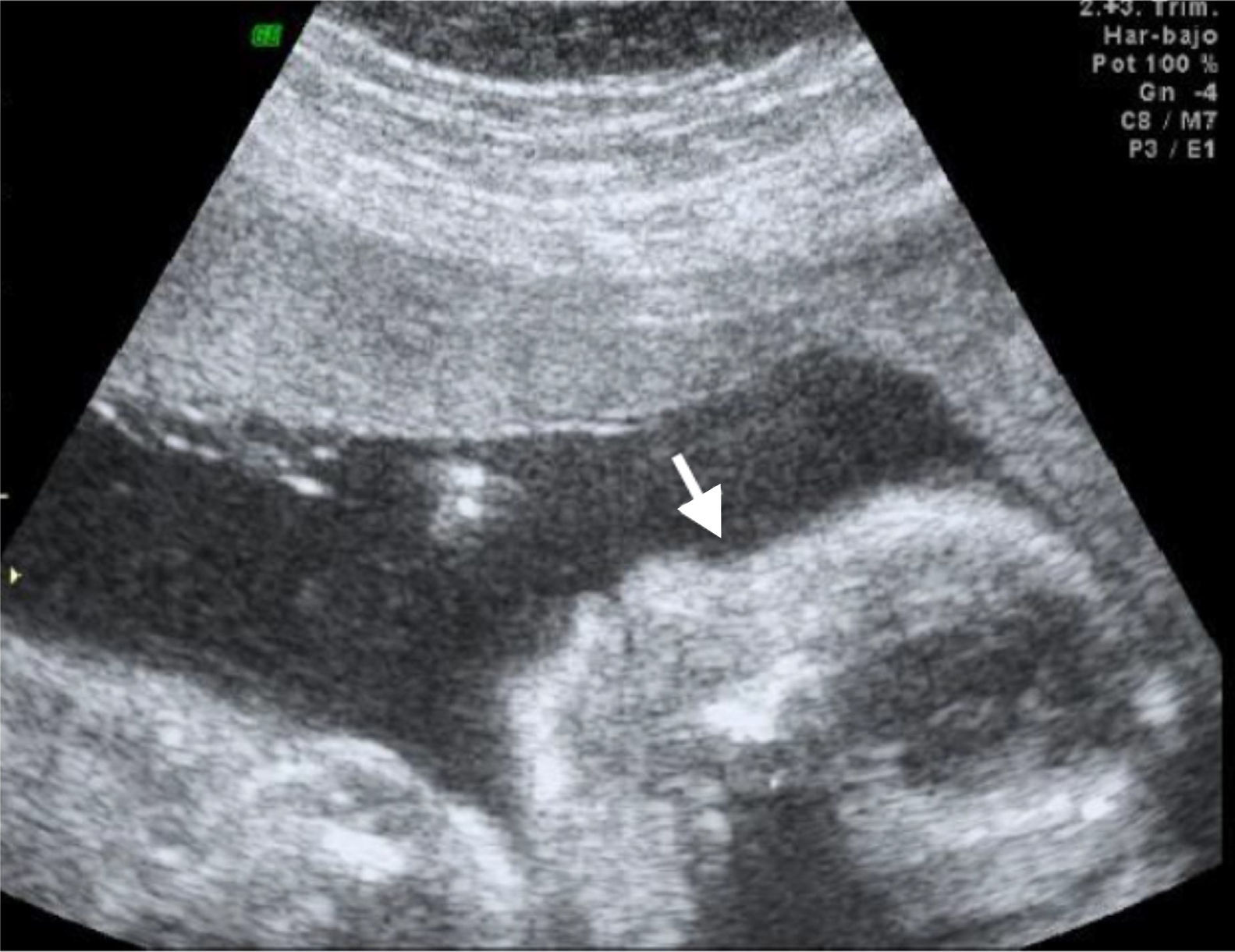
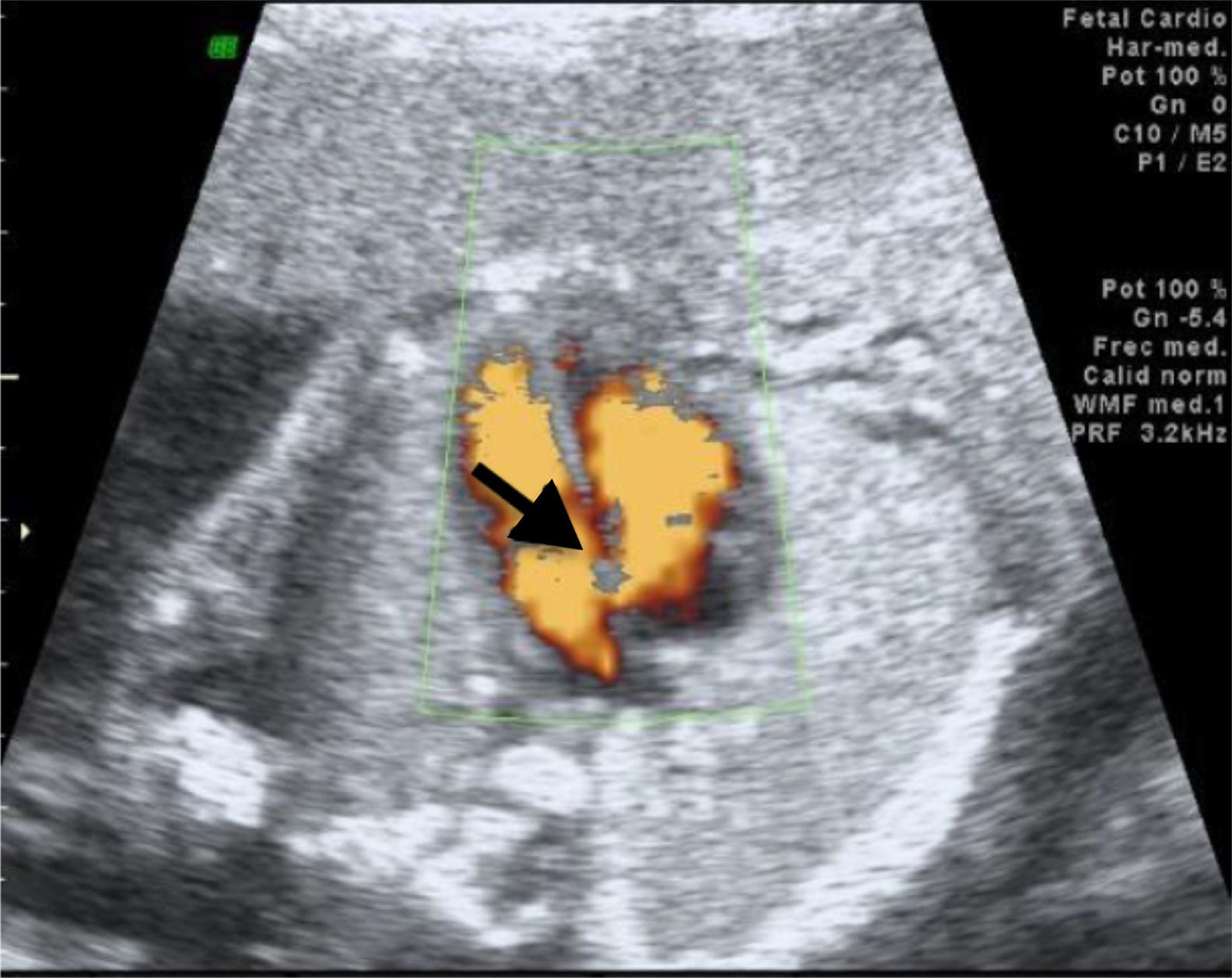
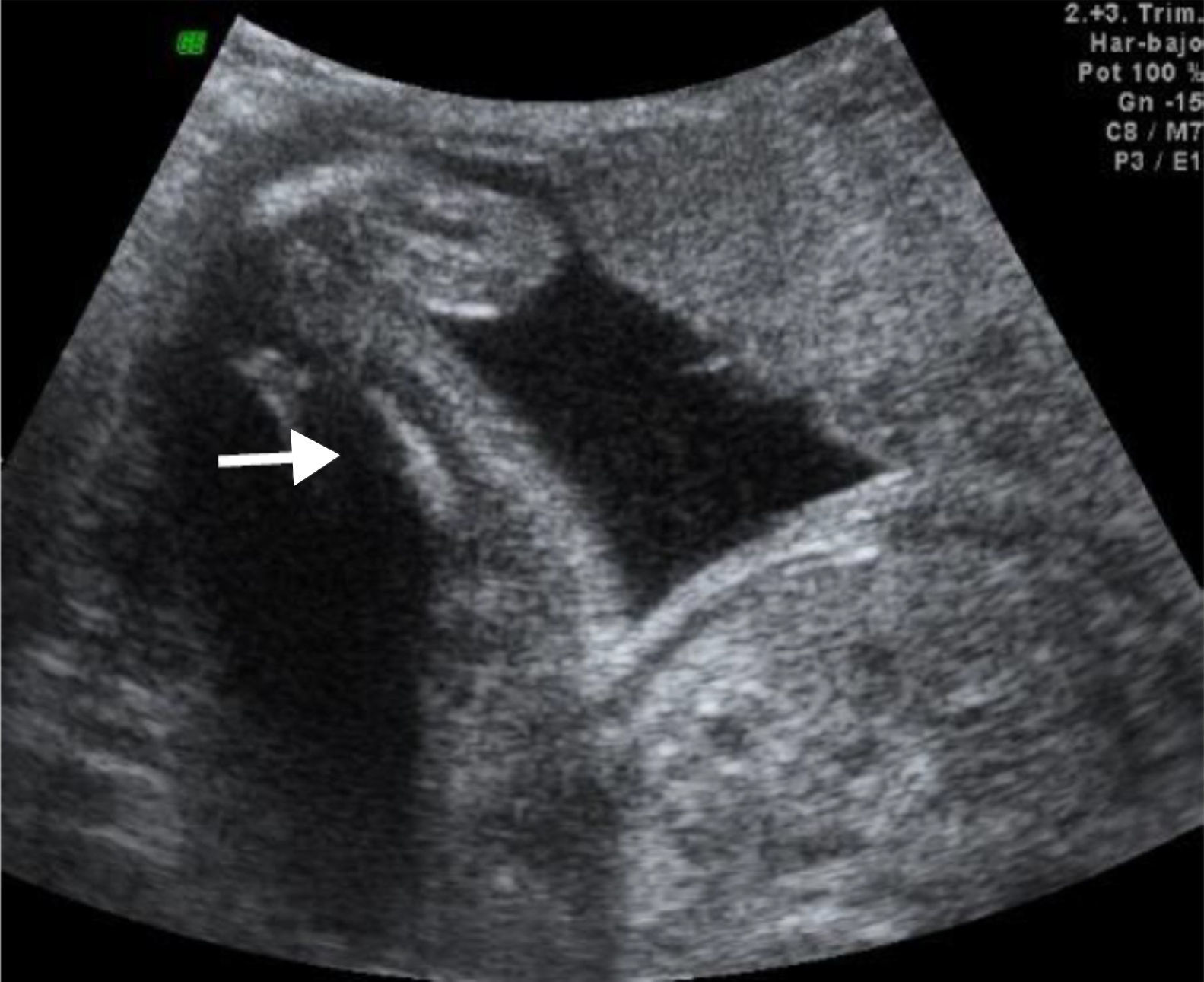
We recently had the opportunity in our state to treat a pregnant patient whose baby was diagnosed with Roberts syndrome, dying a few hours post-C-section. Several anatomical alterations were detected via ultrasound (Figs. 1–3). Although Roberts syndrome is rare, we believe it is appropriate to recapitulate.



Roberts syndrome, also known as pseudothalidomide syndrome or phocomelia syndrome, is a rare pathology of genetic origin with an autosomal recessive pattern, which courses with multiple congenital malformations. It was first described by Roberts in 1919 and defined as a syndrome by Apeltz in 1966. In 1969, Herrmann described it as pseudothalidomide syndrome or phocomelia syndrome, because of the presence of shortened limbs, similar to those observed in embryopathy by thalidomide.1,2
Its incidence is unknown; less than 150 cases are described in the literature to date.1,3 The affected group is diverse and spread around the world. It is characterized by pre- and post-natal growth-delay, microcephalia, a cleft palate and lip, hypoplastic nasal alae, malar hypoplasia, micrognathia, hypertelorism, intracranial aneurisms, capillary hemangioma, exophthalmos, palpebral fissures with a downward slant, dysplastic or small ears, and cataracts or cloudy cornea. Heart congenital anomalies may also occur, such as interventricular communication and patent ductus arteriosus, cystic kidneys, enlarged genitalia, oligodactyly with thumb aplasia or hypoplasia, syndactyly, or clinodactyly.1,3–5 Upper limbs are more affected than lower limbs.9 There is parental consanguinity in 49% of the cases.6,7 The disease is caused by mutations of the ESCO 2 gene (establishment of cohesion of the counterpart 2), from which 26 mutations have been described to date. This gene is located in the short arm of the human chromosome 8p21.1, codifying it as an acetyltransferase involved in the regulation of the sister chromatid cohesion.1,3 Roberts syndrome has been shown to be a “mitotic mutant”.8
At a cytogenetic level, a premature separation of heterochromatin of the centromeres is described, usually most evident in the acrocentric chromosomes and in the long arms of the Y chromosome. On the other hand, the premature separation of the centromeric heterochromatin has only been observed in 50% of the cases.2,6,10–12 The risk of recurrent disease is 25% of couples with a positive family background. Prenatal diagnosis of risky pregnancies requires an ultrasound at 12 weeks gestation; chorionic villus sampling at 8 weeks gestation will be performed in women with a high recurrence risk of Roberts syndrome. Prenatal cytogenetic analysis is conducted in the cells in the chorionic villus samplings, either amniocentesis or cordocentesis. However, a negative cytogenetic analysis does not exclude diagnosis.
Prognosis is adverse. High mortality rate during the neonatal period and childhood are mainly due to cardiac or renal malformations.
Intellectual disability occurs in most of the affected subjects; in some cases, intelligence can be normal. Mildly affected individuals may survive until adult age, and treatment is designed to improve quality of life.
FundingNo financial support was provided.