




Worldwide, the hepatitis C virus (HCV) infection is a leading etiology of cirrhosis and hepatocellular carcinoma (HCC). The main complications of chronic HCV infection are fibrosis, cirrhosis and HCC. Nowadays, there are several options accessible for HCV treatment, such as direct-acting antiviral agents (DAAs), including HCV protease inhibitors, polymerase inhibitors, and NS5A inhibitors. Once the virus is eliminated after the specific antiviral treatments, patients should try to re-establish their metabolic functions efficiently from an environment where there was great cellular destruction. Another goal for the liver can range from fibrosis to decompensate cirrhosis. For this reason, upon the use of DAAs therapy, assuming that SVR is reached, the use of antifibrotic drugs is increasing in order to improve liver regeneration by decreasing fibrotic tissue generated by HCV. Liver fibrosis is defined as the excessive accumulation of extracellular matrix proteins because of the interaction of a number of different cell types, among them hepatic stellate cells. Once the causative agent of damage is removed, the progression of liver disease is attenuated. However, if the patient has an advanced state of fibrosis or cirrhosis, hepatic regeneration becomes a challenge where numerous known and unknown cellular mechanisms are involved. Fortunately, there are drugs that facilitate this process of reversion, however, it is still not clear that conditions favor the success of this regression.
Chronic liver diseases represent a major health problem worldwide, including chronic viral hepatitis, metabolic disorders, malnutrition, alcohol abuse, and autoimmune diseases, and are a major cause of chronic liver injury and subsequent complications, such as liver cirrhosis or HCC.1,2 Hepatitis is an inflammation of the liver characterized by jaundice, abdominal pain, liver enlargement and sometimes fever that may trigger different conditions like nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), or cholestasis. Regarding viral hepatitis, many different viruses, such as hepatitis A, B, C, D, E, F and G, can all cause viral hepatitis.
Worldwide, the hepatitis C virus (HCV) infection is a leading etiology of cirrhosis and hepatocellular carcinoma (HCC). The main complications of chronic HCV infection are fibrosis, cirrhosis and HCC. The interaction of HCV with its human host is complex and multilayered; direct and indirect mechanisms of HCV-induced HCC include activation of multiple host pathways such as liver fibrogenic mechanisms, cellular and survival pathways, interaction with the immune and metabolic systems, and viral genotype, etc.3
Despite the fact that a protective vaccine against HCV is not yet available, there are several options accessible for HCV treatment, like direct-acting antiviral agents (DAAs), including HCV protease inhibitors, polymerase inhibitors, and NS5A inhibitors. Some of them, like sofosbuvir, daclatasvir, ledipasvir, and ombitasvir, among others, are used in HCV infected patients with different genotypes, achieving a sustained virological response (SVR) at the end of the treatment. Nevertheless, in patients with cirrhosis, results remain unclear but show promising responses.4 Ongoing research is focused on improving the pharmacokinetics and tolerability of these agents, finding the best regimens, and developing treatment strategies that produce the best outcomes.5
The livers of the patients who have eliminated the virus after the specific antiviral treatments should try to re-establish their metabolic functions efficiently, from an environment where there was great cellular destruction. The challenges the patients face can range from fibrosis to decompensated cirrhosis. For this reason, upon the use of DAAs therapy, assuming that SVR is reached, the use of antifibrotic drugs such as pirfenidone, farglitazar, irbesartan, and PRI24,6 are increasing in order to improve liver regeneration by decreasing fibrotic tissue generated by HCV. The pursuit of antifibrotic therapy is to avoid the establishment of a cirrhotic state and helps to reestablish normal liver function upon HCV infection.
Hepatitis CHepatitis C virus (HCV) infection is a worldwide pandemic with an estimated 150–200 million people infected globally (2.8%), and there are three to four million people newly infected each year, as estimated by the World Health Organization (WHO).7,8
A diagnosis of HCV infection can be carried out using indirect tests, such as through a detection of anti-HCV antibodies by recombinant immunoblot assays (RIBA) or the use of direct tests such as detection of viral RNA by PCR as well as antigen detection of viral proteins by enzyme immunoassay (EIA).9
It is known, according to the natural history of the disease, that approximately 75–85% of all patients with an acute HCV infection will develop chronic hepatitis C, which is defined as the persistence of HCV RNA over six months. A chronic HCV infection is influenced by several factors, such as age at time of infection, ethnicity, gender and the establishment of jaundice during the acute infection.10
Around 2–30% of subjects infected with HCV will progress to a state of severe liver disease with fibrosis, cirrhosis or HCC over 30 years.11 Nowadays, there are several antiviral regimens that have been approved. In 2011, boceprevir and telaprevir, two first generation NS3/4A protease inhibitors, were authorized by the FDA (Food and Drug Administration) for their use in combination with pegylated interferon (PEG-IFN) and Ribavirin (RBV) for a 24-to-48-week course of treatment in HCV-genotype 1 infections. Two years later, in 2013, simeprevir (a second-generation NS3/4A protease inhibitor) was approved for use with PEG-IFN and RBV for a 12-week course of treatment in HCV-genotype 1. In addition, sofosbuvir (a NS5B nucleotide polymerase inhibitor) was approved for use with PEG-IFN and/or RBV for a 12 or 24-week course of treatment in HCV-genotypes 1 to 4. IFN-free regimens have been shown to give better results, along with sofosbuvir, combined with simeprevir or a NS5A replication complex inhibitor (ledipasvir or daclatasvir), with or without RBV for a 12-week treatment in genotype 1, resulting in a sustained virological response (SVR) over 90%. Nowadays, the international guidelines for HCV treatment recommend the PEG-IFN free regimens.12
The benefit of extending SVR, thanks to antiviral drug efficacy, is to block the fibrogenic progression of chronic liver disease, thus avoiding the progressive destruction of normal tissue architecture or the replacement of hepatic parenchyma. On fibrous tissue; the final outcome of this process is liver cirrhosis, which is the major cause of morbidity and mortality in chronic viral hepatitis.13
The use of DAAs has given us powerful information about the natural history of fibrosis regression; DAAs have established important benchmarks and biological blanks for antifibrotic drugs.
For all previous mentions, it is important to focus the research on really learning about the molecular mechanisms of fibrogenesis in order to develop new drugs and treatments that produce regression of fibrosis regardless the origin of fibrotic disease.14
Liver fibrosisFibrosis is a reversible scarring response that occurs in almost all patients with chronic liver injury from different ethnologies. Generally, hepatic fibrosis leads to cirrhosis, and is associated with nodule formation and organ failure.
The fibrotic response is a consequence of many complications of end-stage liver disease, like portal hypertension, ascitis, encephalopathy, synthetic dysfunction, and impaired metabolic capacity. Due to this background, it is imperative to understand and attenuate fibrosis because it has direct clinical implications in liver disease.15
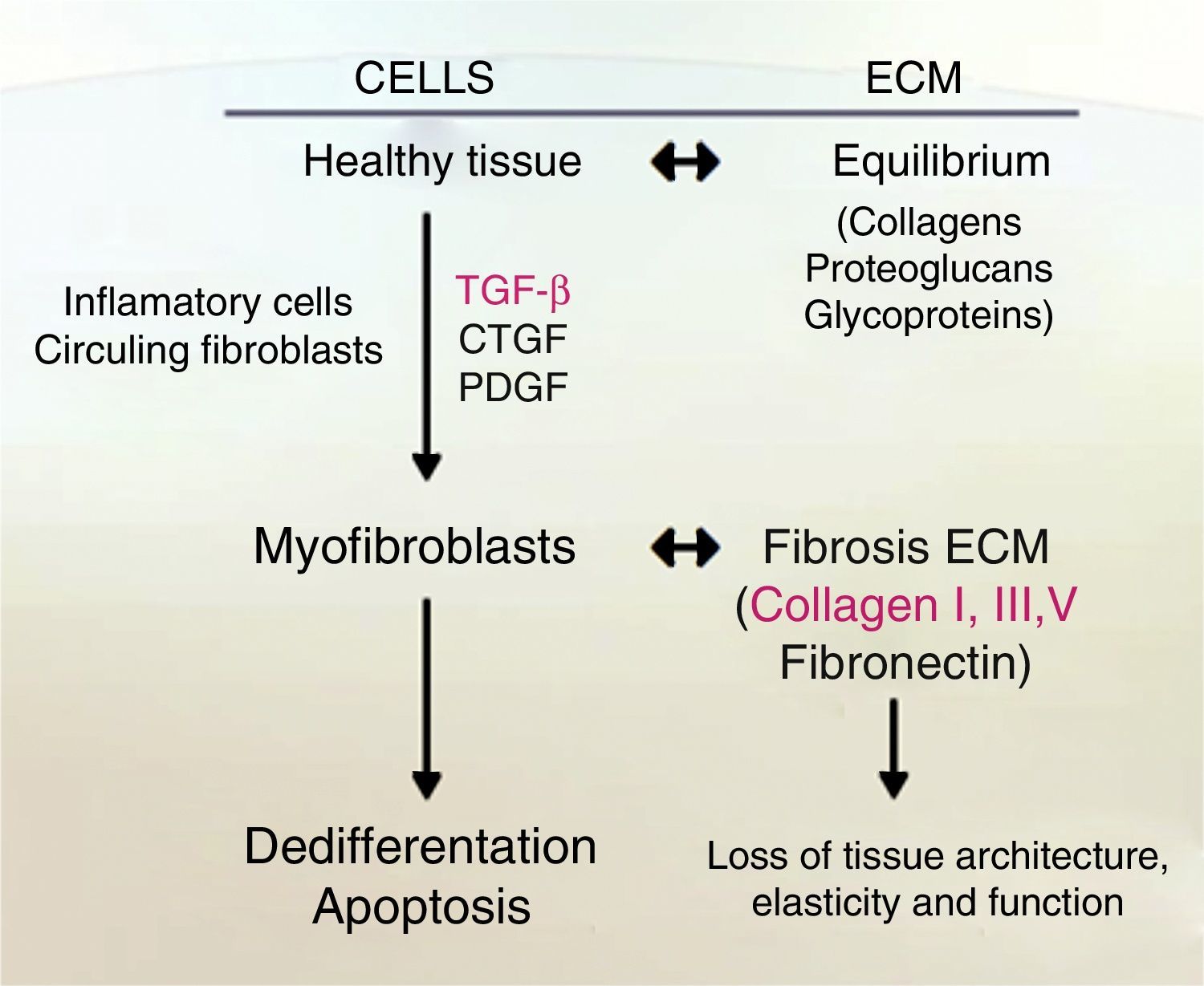
Liver fibrosis is defined as the excessive accumulation of extracellular matrix proteins because of the interaction of a number of different cell types including macrophages, myofibroblasts and epithelial cells, leading to an increase of fibrillar collagens (predominantly collagens I and III).16 Fibrogenesis is a dynamic and complex process, mediated by necro-inflammation and activation of HSC.17
Role of hepatic stellate cells (HSC)In 1876, Karl Kupffer discovered cells that had extensions which had contact with the sinusoidal wall and hepatocytes, which he called “Sternzellen” (stellate cells).18 There are between 5 and 8% of HSC in the total population in a normal liver. HSC are mesenchymal, perisinusoidal cells founded in the Disse space, which is located between the fenestrated endothelium of sinusoids and the hepatocytes.
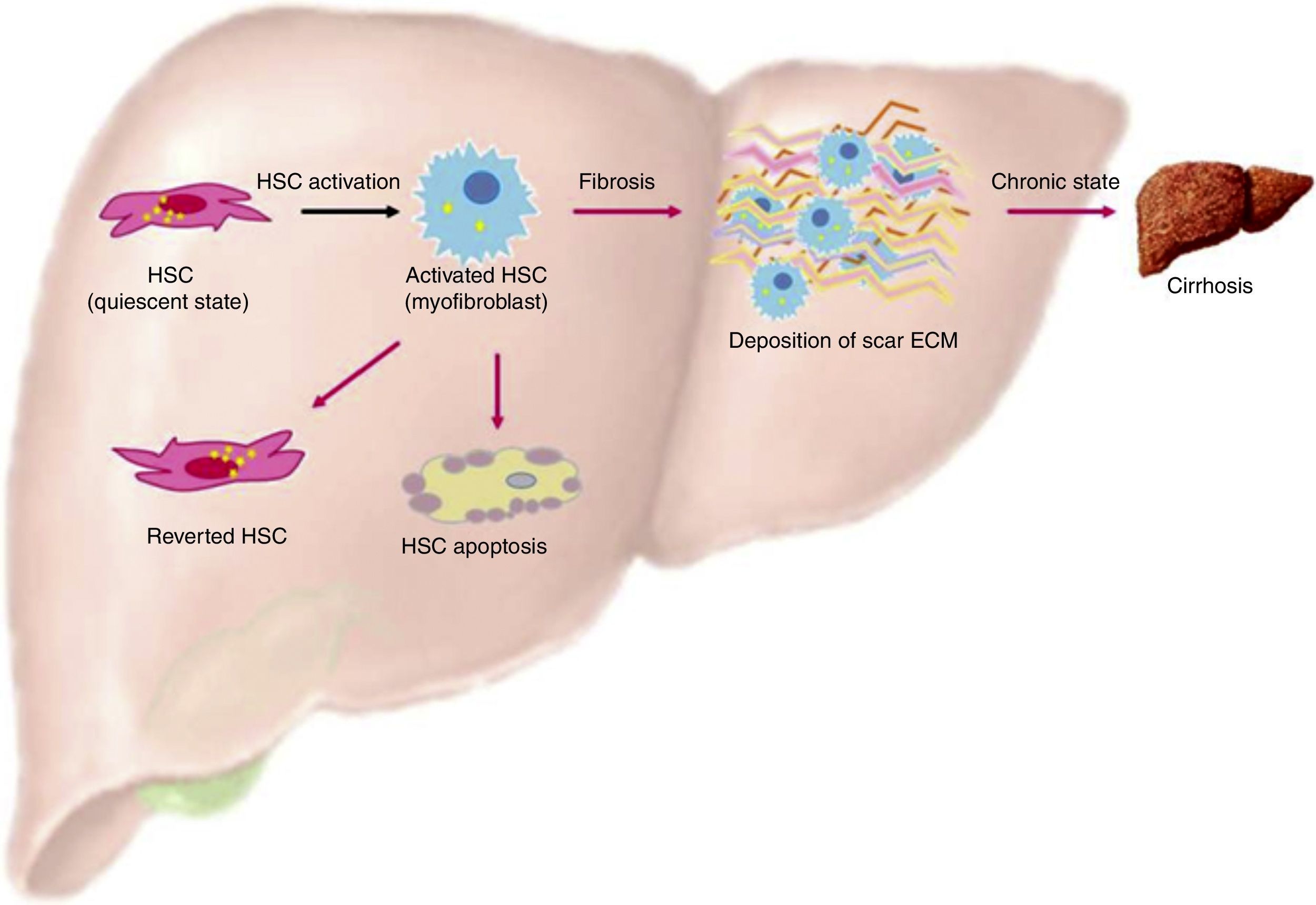
One of the most important characteristics of HSC, when they are in a quiescent state, is the presence of lipid droplets in the cytoplasm containing vitamin A as a retinyl palmitate19 (see Table 1 and Fig. 1).
Functional characteristics of HSC.18
| Characteristics of HSC in normal liver | |
|---|---|
| Functiona | Related molecules |
| Lipids droplets storage | Vitamin A as retinyl palmitate |
| Control of intercellular communication | PDGF, PI-3k, FGF-2, VEGF, TGF-β, ET-1, ET-2, ET-3, angiotensin-II, vasopressin, thrombin, MCP-1, TNF, IL-1, IFN-γ.b |
| Remodeling of ECM | Collagen, MMPs and TIMPs |

Activation of HSC. This is a general view of HSC activation, transforming them into myofibroblast phenotypes, increasing production of ECM in the liver and stress in liver tissue. On the other hand, another ending for HSC is apoptosis, where overproduction of ECM by these cells is finished. Finally, HSCs can actually revert from their myofibroblast phenotypes into a quiescent state, but these can easily change into a myofibroblast phenotype again.
When liver injury is established, HSC lose their lipid droplets and undergo important morphological and functional changes, acquiring a myofibroblast-like cell phenotype and beginning overproduction of collagen, mainly type I; this process is known as activation.19
One indicator of activated HSC is the expression of α-smooth muscle actin (α-SMA), an actin isoform which is not present in other liver cells, other than the smooth muscle cells surrounding large vessels. Moreover, activated HSC also expresses other marker genes that are present in smooth muscle cells too, for example, smooth muscle myosin heavy chain, calponin and myocardin. Nevertheless, activated HSC still differs from myofibroblasts and smooth muscle cells in their vitamin content, contractile activity, and their susceptibility to some cytokines, specially to transforming growth factor-β1 (TGF-β1) (see Fig. 2).19–22

Fibrosis, in some cases, is reversible; it can be said that in this disease, the main characteristic is the disordered accumulation of MEC fibrillar type proteins (mainly collagen) which reduces the fenestrations between the sinusoidal cells in order to eliminate them and decrease liver function, with a loss of normal architecture and nodule formation regeneration.23
In addition, sinusoidal cells increase the expression of pro-inflammatory molecules including intercellular adhesion molecule-1 (ICAM-1), vascular endothelial cell growth factor (VEGF) and adhesion molecules,24 presenting a neoangiogenesis and contraction of scar tissue leading to elevated tissue tension.14
Accumulation of fibrillar proteins is the outcome of an imbalance between ECM synthesis and degradation plus to a low production of MMPs (Matrix metalloproteinases), or their inhibitors overproduction, TIMPs (tissue inhibitors of metalloproteinase), or pro-fibrogenic cytokine as TGF-β.23,25
Role of metalloproteinasesIt is known that metalloproteinases help the remodeling of ECM because of their catabolic function over different components of this matrix. There are several MMPs in the human body, but it is necessary to know the principal mediators in liver fibrosis. There is metalloproteinase-1 (MMP-1), which performs interstitial collagen lysis; its activation depends on metalloproteinase-3 (MMP-3); it has a molecular weight of 55kDa as zymogen and 45kDa as an active form. Metalloproteinase-9 (MMP-9) breaks gelatins; MMP-3 is one of the few MMPs that is not specific to a single compound, as it degrades laminin, fibronectin, proteoglycans, collagen, and gelatin. The main cellular source of MMP-3 is HSC. It can activate MMP-1 and MMP-9, which are very important for the degradation of MEC proteins during fibrosis. MMPs can be classified according to their affinity for a substrate. Although very specific in their degradation activity, they have several structural characteristics, such as having been secreted into the interstitial space of cells in a latent shape. In Table 2 we show some MMPs and their specific substrates.26
When liver cirrhosis is present, fibrillar collagen type I and III are deposited in the liver lobules25 forming irregular septa, which can become very thick and divided, surrounding portions of the liver lobule (regenerative nodules).27 This protein-like material has the consequence of recruiting inflammatory cells. The HSC are the cells responsible for the production of almost all collagen in ECM. HSC suffer a radical change called activation, which consists of several stages, changes into a myofibroblastic phenotype, proliferates and become a strong producer of collagen type I and III, TIMP-1, TIMP-2 and TGF-β, losing its main function of storing 80% of the total vitamin A in the whole body as retinyl palmitate in lipid droplets in the cytoplasm, and the regulation of transporting and storing vitamin A.28
By the early stages of HSC activation, there is a pre-inflammatory state, there are some changes in gene expression, and the cell becomes sensitive to some cytokines like TGF-β.29
At this state of fibronectin production via TGF-β, the infiltration of Kupffer cells, stimulates ECM synthesis and the release of retinoids by the HSC, losing their principal function; besides the production of ECM. HSCs increase in number. This stimulation is also directed by a mitogenic cytokine known as platelet-derived growth factor (PDGF).26
In fibrogenesis, ECM proteins are overexpressed by TGF-β; different types of MMPs, TIMP-1 and TIMP-2, lead to a replacement of molecular collagen types.30
Pathogenesis of liver fibrosisMechanisms involved in the production of an extracellular matrix in hepatic fibrosis have had great advances that have helped in the elucidation of the stages of fibrosis and future therapies. During liver fibrosis and cirrhosis, there is an increasing accumulation of high density fibrillar collagens (for example, collagens I and III), as well as proteoglycans and other constituents of the ECM.23,24,31
In the progression of fibrosis in a chronic state, the cells of the liver experience different changes according to their cell type. Damage hepatocytes undergo apoptosis. On the other hand, the sinusoidal endothelial cells change to a state of acapillarization of the sinusoids. The Kupffer cells activate and immediately produce different chemokines and cytokines. Lymphocytes infiltrate the areas of the liver that present damage and increase the inflammatory process. In addition, the quiescent HSCs change their status to an activated state, increasing ECM.32
Liver fibrosis regressionThere is now solid evidence of reversion of activated stellate cells to a more quiescent state in rodent models of fibrosis.33–35 Three major mechanism in the resolution of fibrosis have been found: one of these is HSC becoming apoptotic, another is HSC becoming senescent, and finally there is HSC reversing their phenotype to an inactivated state.25,35
HSC and apoptosisExperimentation in rat models has shown that the recovery phase after 4 weeks of treatment with CCl4 is associated with decreased TIMP-1 in its early stages, as well as a decrease in the density of activated HSCs by apoptosis.36 Experiments in transgenic mice with TIMP-1 scavengers demonstrated the causal relationship between hepatic expression of TIMP-1, fibrolysis failure, and increased survival of HSC.16,25 Another study identified NF-κB as an important transcription factor in the upregulation of anti-apoptotic genes; however, the signaling pathways activated by CCl4 are different from those triggered when an HCV infection is present. It would be ideal to compare these results, as well as the half-life of the HSC-activated in vivo model, with different initiators, simulating fibrosis liver damage (see Fig. 1).
HSC becoming senescentSenescent HSCs can contribute to the regression of fibrosis, since it no longer presents cell division, and they increase the expression of enzymes responsible for the degradation of ECM.37
Moreover, senescent hepatic myofibroblasts can be eliminated by natural killer (NK) cells. Thus, senescent hepatic myofibroblasts may prevent the proliferation of these cells producing ECM, promoting ECM degradation and accelerating clearance of myofibroblasts at the injury site.38
Reversion of HSC phenotypes to an inactivated stateRecent studies in in vitro cell tracking have shown that activated HSCs can be disabled and display a quiescent phenotype after cessation of liver damage.33,34,38
However, the reverse HSCs do not have all the features of quiescent cells, but rather maintain an intermediate activated state with increased susceptibility to fibrogenic stimulus.33,34
These data raise the possibility that reverted HSCs can contribute to the reversion of fibrosis, but may promote a rapid progression of fibrosis and a severe time of recurrence of liver injury.25
Drugs with antifibrotic and anti-inflammatory effectsPFD (pirfenidone) has proven anti-fibrotic and anti-inflammatory properties in animal models of fibrosis.39 PFD's effects are mediated in part through inhibition of NF-κB protein activation.40 Also, PFD down-regulates TGF-β1, TIMP-1, MMP-2 mRNA and collagen deposition in an experimental rat model of cirrhosis induced by CCl4.39,41
PFD has effects on necroinflammation, fibrosis and steatosis, and was able to decrease serum levels of TGF-β1, IL-6, TNF-α and hepatic CB1, as well as gene expression of CB2 in patients with HCC. Additionally, PFD inhibited TCR-induced production of multiple proinflammatory cytokines and chemokines.42
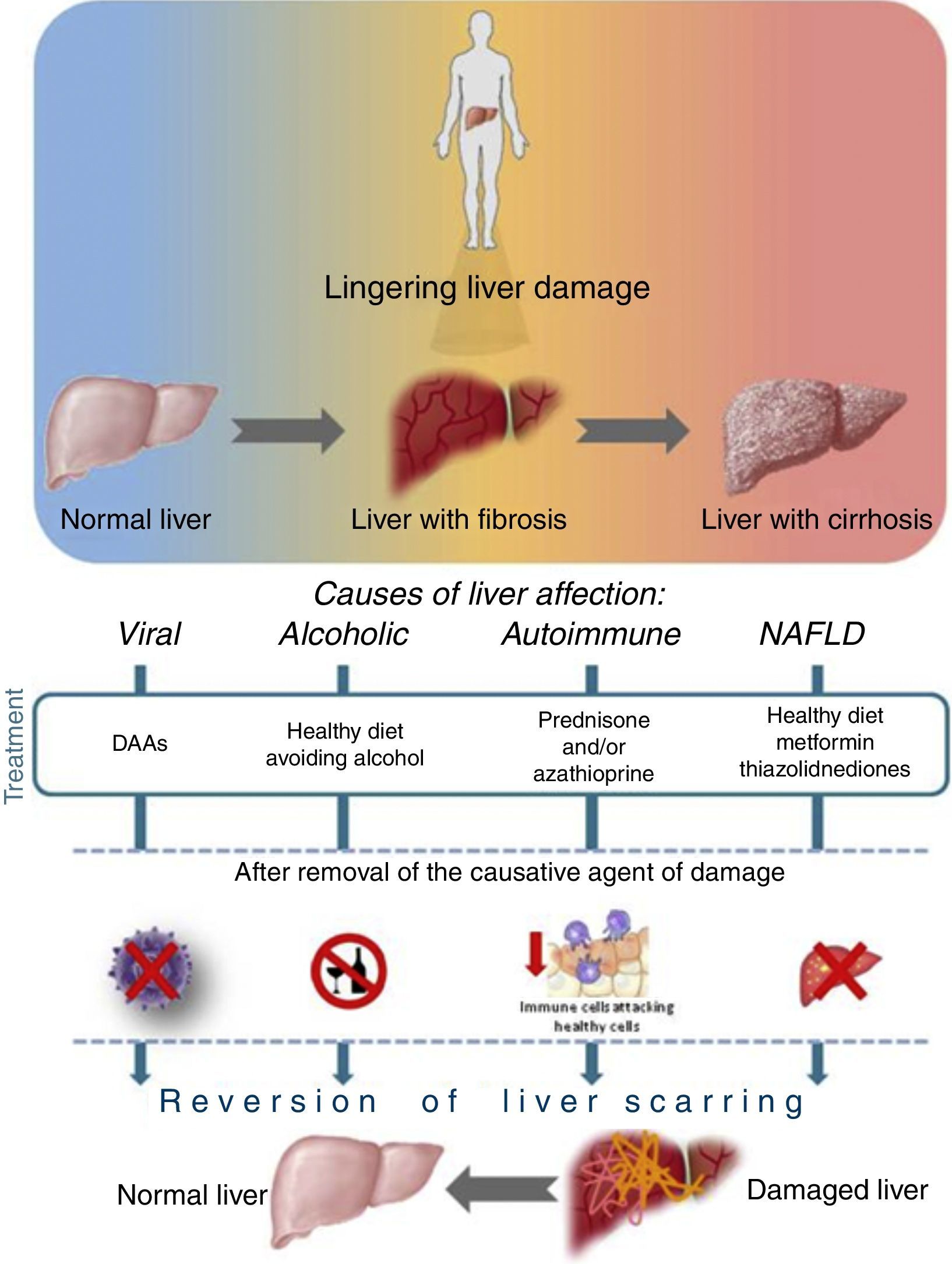
Liver recovery after different etiology of damageAs is well-known, once the causative agent of damage is removed, the progression of liver disease is attenuated, and recovery is a possible scenario even if the patient has an advanced state of fibrosis or cirrhosis. See Fig. 3.

The DAAs for patients with HCV infection in an advanced fibrosis stage have revolutionized treatment choices. The patients have developed high response rates with compensated cirrhosis in clinical trials,43–46 which has been observed even in patients with decompensate cirrhosis, for whom Peg-IFN is contraindicated and who in other ways would not have a good prognosis; at this time, HCV can now be eliminated.4,47
One of the main goals in the regression of fibrosis is to redirect the synthesis of inflammatory molecules to a restorative state. For this, healthy hepatocytes and non-parenchymal cells change the microenvironment from a pro-inflammatory ambience (environment) to resolution, so that the presence of anti-inflammatory mediators increases. The liver macrophages become a restorative phenotype, increasing the expression of MMP and growth factors leading to hepatocyte recovery. In liver fibrosis, regression is increased, as is the number of dendritic cells (DCs) and NK cells in the liver. DCs favor matrix degradation via synthetic activation of MMP-9. Another cell type involve in regression are the NK cells, which induce the apoptosis of activated and senescent myofibroblasts via NKG2D and TRAIL.48
On the other hand, it is important to eliminate the excessive extracellular matrix: the MMPs play a very important role in this stage. The objective of some macrophages in the liver microenvironment is to release some anti-inflammatory mediators, as well as provide such fibrolytic mediators, like MMP12 and MMP13. Nevertheless, neutrophils and HSC can be expressed in other MMPs. But some characteristics of advanced fibrosis adjudge some resistance to matrix degradation; these features are collagen cross-linking and deposition of elastin, among others.48,49
AlcoholThere are no FDA-approved treatments for alcoholic liver disease (ALD), which ranges from simple steatosis to cirrhosis and HCC. Abstinence from alcohol leads to a resolution of alcoholic fatty liver disease (AFLD). It is a benign steatosis, besides improving survival in a cirrhotic state even in those patients with decompensated liver function. There are some off-label pharmacotherapies aimed at improving the quality of life of patients with ALD. These tend to involve the use of glucocorticosteroids,50 and pentoxifylline,51 among others.52
Autoimmune hepatitisAutoimmune hepatitis (AIH) is a chronic inflammatory liver disease, which, if untreated, can develop into cirrhosis, liver failure or even death. There is not yet a medical cure for this condition, but current drugs can control autoimmune hepatitis. The pharmacotherapy acts by suppressing or slowing down an overactive immune system. There are reports where certain treatments may reverse some of the scarring in the liver. These treatments include prednisone and/or azathioprine; these drugs help suppress the immune system and reduce inflammation in the liver.53
NAFLDIn NAFLD, the patient presents hepatic steatosis without hepatocellular injury of hepatocytes, which could have the shape of a balloon. The presence of hepatic steatosis cannot be secondary to hepatic fat accumulation like alcohol consumption, use of steatogenic medication or hereditary disorders. The management of patients with NAFLD consists of treating the liver disease, as well as metabolic co-morbidities such as obesity, hyperlipidemia, insulin resistance and type 2 diabetes mellitus (T2DM), leading to results like decreasing fat, fibrosis and necroinflammation with metformin, or a larger weight loss and reversal of steatosis with Orlistat, all therapies depending on the type of patient and the state of the disease.54
At the moment, there is no exhaustive information about the extent of the hepatocyte's ability to regain function and hepatic metabolism (periportal to perivenous differentiation) within the hepatic lobule after SVR. It is important to consider the differences between cirrhosis and fibrosis, since in many patients it has been reported that this depends on the possible reversibility of the fibrotic state. During fibrosis stages, the amount of collagen increases, as does the ratio of fibro-connective tissue and liver cellular tissue, but the liver lobular structures are unbroken, and there is no pseudo-lobule formation. Cirrhosis consists of two pathological features: fibro-connective tissue hypertrophy and pseudo-lobule formation. At the cirrhosis stage, the liver's essential structure is damaged, and liver structure begins to collapse. Thus, reversal is more difficult at this stage; for this reason it is important to consider the severity of the liver disease. A study reported patients with decompensate cirrhosis showed an SVR of nearly 90% with a DAA regimen. This study also reported on recovery in serum markers of synthetic liver function for a majority (>50%) of patients at post-treatment week 4. For this reason, it is important to analyze if an improvement in the clinical tests is presented over time.55,56
However, if we prolong the follow-up of patients, not all patients with high rates of fibrosis will return the same results, so whether or not there is a point of no return in fibrosis is still up for debate; it can be said that the cause might be structural. Another point to be discussed is that there are no official standardized studies addressing regressed fibrosis, so it remains that this parameter should be taken in mind when reporting results. However, there are several tools and markers that can help in the evaluation of fibrosis regression, like ultrasound-based transient elastography (TE) FibroScan® (Echosens, Paris, France) and FibroTest® (Biopredictive, Paris, France; licensed as FibroSure® in the USA), and of course a blood-based biomarker.57,58
Among the benefits of being able to eradicate the causative agent of hepatitis C, and as a consequence the continuous hepatic injury, is that the patient can increase his quality of life, as well as prevent a condition that requires liver transplantation and its consequences. On the other hand, extrahepatic comorbidities, like mixed cryoglobulinemia vasculitis, renal disease, type II diabetes mellitus, cardiovascular disease, porphyria cutanea tarda, lichen planus, and lymphoproliferative disorders such as B-cell non-Hodgkin lymphoma, irritability, malaise and other neuropsychiatric symptoms like depression, can all be avoided.57
ConclusionThe present work summarizes how the liver manages different types of damage, and the result of its resolution. Focusing on HCV, one of the principal goals in fibrosis regression is to redirect the synthesis of inflammatory molecules to a restorative state. For this, healthy hepatocytes and non-parenchymal cells are needed to change the microenvironment from a pro-inflammatory ambience to an ambience of resolution. On the other hand, the use of antifibrotic drugs during treatment against HCV helps avoid the increase of ECM deposition. This way, the quantity of HSC decreases. This field of liver regression and restoration of liver damage promises a better future for patients who resolve HCV disease in an advance stage of fibrosis or cirrhosis.
Conflict of interestThe authors certify that there are no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.