



Reversible posterior leukoencephalopathy syndrome (RPLS) is a clinical/radiological syndrome with numerous possible causes. This condition is increasingly recognised due to the rising use of neuroimaging techniques, although its incidence remains unknown. All age groups are susceptible.1 It is more frequent in women, even when patients with eclampsia are excluded.2 It is often associated with several clinical situations: severe arterial hypertension (AHT), eclampsia, kidney disease, immunosuppressant treatment, autoimmune disease, blood transfusion, and contrast exposure. The most frequent symptoms are headache, altered level of consciousness, visual alterations, and seizures.3 Cranial MRI reveals vasogenic oedema in the cortical and subcortical white matter, typically limited to the parieto-occipital region of both hemispheres, although several other localisations have been reported (frontal lobe, temporal lobe, basal ganglia, cerebellum, and brainstem).3,4 Pathogenesis remains unclear. Symptoms and haemodynamic alterations fully resolve when the underlying cause is corrected promptly, and although neuroimaging findings resolve more slowly (weeks to months),5 follow-up neuroimaging studies with specific sequences are needed to confirm diagnosis.1
We present the case of a 79-year-old woman who, following a fall, presented disorientation, incoherent speech, and mild-moderate left hemicranial pulsatile headache of severe intensity accompanied by photophobia/phonophobia with no nausea or vomiting, which had progressed for 7-10 days. She showed no head trauma, altered level of consciousness, sphincter incontinence, fever, psychiatric history, or toxic habits. Her medical history included AHT, congestive heart failure, gastric ulcer, sigmoid colectomy 6 years previously (high-grade dysplasia), and no previous chemotherapy. The patient was illiterate and presented a 2-year history of progression-free mild cognitive impairment (MCI). A head CT scan performed one year previously showed mild, predominantly frontal leukoaraiosis. She was receiving treatment with 20mg omeprazole, 40mg furosemide, 120mg diltiazem, and 0.5mg risperidone. At admission, her arterial blood pressure was 200/78mm Hg and heart rate was 65 bpm. Axillary temperature was 37.4°C, and oxygen saturation was 98%. Cardiopulmonary auscultation revealed systolic murmur (grade II/III) and abdominal examination identified no abnormalities. No oedema was observed in the lower limbs. During the neurological examination, the patient was awake but disoriented in time and space, and showed periods of logorrhoea but no agitation. Eye fundus examination revealed no abnormalities. She showed left central facial paralysis and mild dysarthria. No motor impairment or meningeal signs were observed.
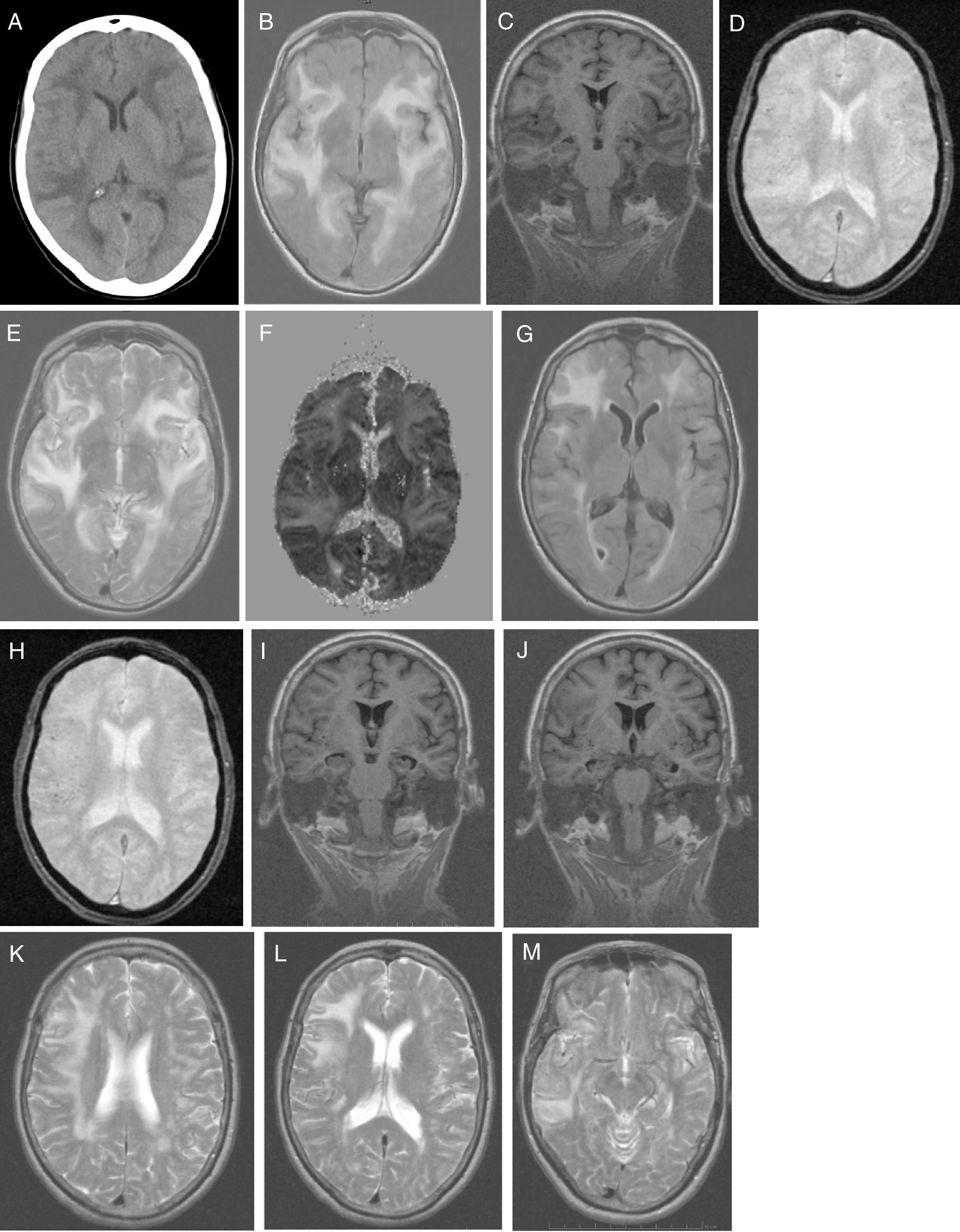
No alterations were observed in the analytical study, which included: blood count; C-reactive protein; erythrocyte sedimentation rate; coagulation; serum electrolyte test; B-type natriuretic peptide; kidney, liver, and thyroid function; vitamin B12; folic acid; autoimmunity; anti-Treponema pallidum antibodies; and HIV types 1 and 2. Results from the urine toxicology test were negative. A chest radiography displayed no abnormalities. A head CT scan (Fig. 1A) revealed large asymmetrical, confluent hypodense areas in both hemispheres, which were apparently oedematous, predominantly subcortical, and mainly affected the left side; these lesions are suggestive of RPLS, which is better studied by brain MRI. We started treatment with oral captopril dosed at 25mg every 8h, intravenous furosemide at 20mg every 6h, and paracetamol for 5 days. Clinical progression was excellent (speech was coherent and headache resolved, but the patient remained disoriented in time) and the patient was haemodynamically stable. Five days after symptom onset, a brain MRI scan (Fig. 1B-F) revealed a large, confluent hyperintensity (frontal, temporal, parietal, occipital, and insular regions in both hemispheres) surrounding the white matter (finger-shaped lesion) and cortex; few juxtacortical hypointense foci (microhaemorrhages) were observed. T2-weighted and FLAIR sequences revealed ischaemic gliotic foci in the white matter. The ADC map showed increased signal with no diffusion restriction, suggesting RPLS, which is less frequent than toxic-metabolic encephalopathy. The patient was discharged on treatment with 160mg valsartan and 12.5mg hydrochlorothiazide, with well-controlled blood pressure. Four weeks after symptom onset she was examined in the outpatient clinic; her Mini–Mental State Examination score was 14 (4 in orientation, 3 in registration, 3 in attention and calculation, 0 in recall, and 4 in language and copying) and functional disability with preserved basic activities of daily living, with difficulties in instrumental tasks involving food preparation, laundry, transportation, responsibility for own medications, and finances. A genetic study confirmed APOE ε4/ε4 genotype. The patient started treatment with extended-release galantamine at 8mg; treatment was discontinued due to poor tolerance (abdominal pain). The patient remained stable with transdermal rivastigmine at 4.6mg. At 2 months after symptom onset, we requested a follow-up brain MRI scan (Fig. 1G-M), to monitor RPLS progression, and a brain PiB-PET scan (Fig. 2), to rule out Alzheimer disease (AD). A brain MRI scan (Fig. 1G-M) revealed less extensive hypersintensity in the frontal, temporal, and parietal regions with no insular or occipital alteration. We observed microhaemorrhages in the frontotemporal region bilaterally on T2*-weighted sequences and ischaemic gliotic foci on T2-weighted sequences. Medial temporal lobe atrophy was also observed; these alterations are suggestive of regressing RPLS.
 Head CT scan: extensive, asymmetrical, confluent hypodensities, apparently oedematous, predominantly affecting the left subcortical region. Bilateral holohemispheric pattern. Baseline brain MRI scan: B) axial T1-weighted sequence: large, confluent, bilateral holohemispheric hyperintensity involving the white matter and cortex; C) coronal T1-weighted sequence: no hippocampal atrophy; D) axial T2*-weighted sequence: few juxtacortical foci of hypointensity suggestive of brain microhaemorrhages; E) axial T2-weighted sequence: hyperintensity in the frontal operculum and bilateral temporal insular cortex extending towards the parietal region on the right and towards the medial temporo-occipital region on the left; F) ADC map: holohemispheric increased signal bilaterally. Follow-up brain MRI scan: G) axial T1-weighted sequence: bilateral fronto-termporal hyperintensity, less marked in the left hemisphere, with no insular or occipital involvement; H) axial T2*-weighted sequence: juxtacortical microhaemorrhages in the fronto-temporal region bilaterally; I-J) coronal T1-weighted sequence: medial and Sylvian temporal atrophy. Axial T2-weighted sequence: K) right holohemispheric hyperintensity of lesser extension; L) right fronto-parietal hyperintensity; M) right temporal hyperintensity.")
A) Head CT scan: extensive, asymmetrical, confluent hypodensities, apparently oedematous, predominantly affecting the left subcortical region. Bilateral holohemispheric pattern. Baseline brain MRI scan: B) axial T1-weighted sequence: large, confluent, bilateral holohemispheric hyperintensity involving the white matter and cortex; C) coronal T1-weighted sequence: no hippocampal atrophy; D) axial T2*-weighted sequence: few juxtacortical foci of hypointensity suggestive of brain microhaemorrhages; E) axial T2-weighted sequence: hyperintensity in the frontal operculum and bilateral temporal insular cortex extending towards the parietal region on the right and towards the medial temporo-occipital region on the left; F) ADC map: holohemispheric increased signal bilaterally. Follow-up brain MRI scan: G) axial T1-weighted sequence: bilateral fronto-termporal hyperintensity, less marked in the left hemisphere, with no insular or occipital involvement; H) axial T2*-weighted sequence: juxtacortical microhaemorrhages in the fronto-temporal region bilaterally; I-J) coronal T1-weighted sequence: medial and Sylvian temporal atrophy. Axial T2-weighted sequence: K) right holohemispheric hyperintensity of lesser extension; L) right fronto-parietal hyperintensity; M) right temporal hyperintensity.
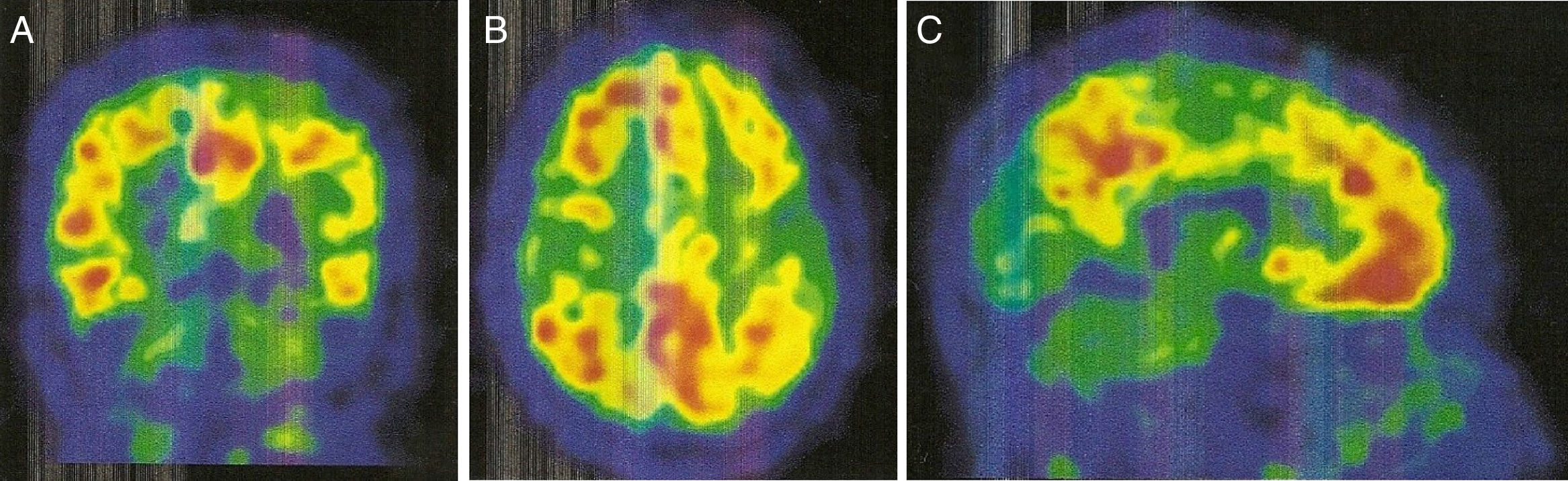
 Coronal slice. B) Axial slice. C) Sagittal slice. Increased radiopharmaceutical uptake in the frontal lobe, superior parietal lobule/precuneus, posterior cingulate, and lateral temporal lobe of both hemispheres.")
An 11C-PiB-PET scan (Fig. 2) showed increased radiopharmaceutical uptake in the frontal lobe, superior parietal lobule/precuneus, posterior cingulate, and lateral temporal lobe of both hemispheres. This finding is compatible with increased cortical beta-amyloid peptide levels.
Six months after admission, the patient was transferred to a day centre and remained stable.
The following scales were applied in the neuropsychological assessment: the Wechsler Adult Intelligence Scale (third edition) and the Wechsler Memory Scale (third edition). Minor alterations were identified in the following variables: attention, immediate auditory memory, episodic memory, vocabulary, psychomotor speed, working and operational memory, verbal intelligence quotient, and learning.
In this clinical case, age of onset was unusually late (79 years). This extensive pattern of bilateral holohemispheric white matter damage in a patient with MCI has not been described in the literature and has not been associated with RPLS, although it is more frequent in transplant recipients, patients taking immunosuppresants, and in cases of autoimmune disease.6
Differential diagnosis of a patient with RPLS and cognitive impairment should include cerebral amyloid angiopathy (CAA). Reversible leukoencephalopathy associated with CAA typically affects posterior regions predominantly7; furthermore, CAA may be a susceptibility factor, with such factors as inflammation and perhaps AHT potentially leading to disease progression.8 The literature has reported cases of rapidly progressive dementia accompanied by seizures, leukoencephalopathy, and mass effect, revealing CAA with fatal outcomes, and a case of CAA with reversible leukoencephalopathy and dramatic improvement of dementia.8
Regarding PiB-PET findings, PiB uptake is moderate in CAA, but with a relative increase in occipital uptake compared with AD; this factor is associated with the distribution of amyloid material in CAA.9,10 Furthermore, PiB-PET shows potential for predicting future haemorrhages; new haemorrhages associated with CAA will mainly appear in sites of increased amyloid deposition.9
Although progression to dementia may have been predictable in the case reported (an elderly patient with vascular risk factors, illiteracy/MCI, and APOE ε4/ε4 genotype), the most probable hypothesis is that RPLS triggered AD; there was no lobar haemorrhage, and cortical microhaemorrhages were scarce. The arguments supporting the diagnosis of AD are: progressive cognitive impairment with functional disability, topographical and neuronal degeneration markers (medial temporal lobe atrophy), a pathophysiological marker (PiB-PET showing increased radiopharmaceutical uptake bilaterally in areas typical of AD), and presence of APOE ε4/ε4 genotype (an AD risk factor).
Homozygosity for ε4 is known to be responsible for younger age of onset of AD,11 and increases its incidence by 9.4 times.12 Another important factor is that the ε4 isoform is more frequently associated with the late-onset and sporadic forms of AD.13
In our case, the presence of the APOE ε4/ε4 genotype did not affect age of disease onset.
Sporadic cases of persistent cognitive impairment have been published in the context of typical RPLS,14 but no cases of AD triggered by bilateral holohemispheric RPLS have been reported.
The pathophysiology of this clinical picture is probably associated with the loss of cerebrovascular autoregulation, causing hyperperfusion and leading to rupture of the blood-brain barrier with ingress of fluid and blood breakdown products to the brain parenchyma.15 Focal cerebral vasoconstriction mediated by brain ischaemia is improbable due to the extension of the oedema and the absence of signs of ischaemia. Endothelial dysfunction has been reported in cases associated with preeclampsia, use of cytotoxic drugs, and autoimmune disease.1,2
This clinical case is unusual in that MCI progressed to AD after correction of AHT in the context of pronounced RPLS, in a patient with APOE genotype ε4/ε4. With this case report, we aim to raise clinicians’ awareness of the early identification of this syndrome, early treatment to avoid neurological sequelae, and the necessary close monitoring of all patients with RPLS through a timely cognitive/neuropsychological assessment.14 Based on the supposed reversibility of RPLS, there is a risk of underestimating a cognitive disorder and failing to adjust treatment.
Please cite this article as: Ros Forteza FJ. Enfermedad de Alzheimer precipitada por el síndrome de leucoencefalopatía posterior reversible. Neurología. 2020;35:279–282.
This study was presented in poster format at the 69th Annual Meeting of the Spanish Society of Neurology (2017).

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas