



El síndrome de leucoencefalopatía posterior reversible (LPR) es un síndrome clínico-radiológico de causa heterogénea. El reconocimiento de esta entidad está aumentando por el uso creciente de las técnicas de neuroimagen, aunque su incidencia es desconocida. Todos los grupos de edad son susceptibles1. Es más frecuente en la mujer, eventualmente cuando son excluidas las pacientes con eclampsia2. Es comúnmente asociado a varias situaciones clínicas: HTA severa, eclampsia, enfermedad renal, tratamiento inmunosupresor, enfermedad autoinmune, transfusión sanguínea y exposición a contraste. Los síntomas más comunes son cefalea, alteración de la consciencia, alteraciones visuales y convulsiones3. La RM craneal revela un edema vasogénico en la sustancia blanca (SB) subcortical y cortical, típicamente limitado a la región parieto-occipital de ambos hemisferios, aunque también se han descrito en otras localizaciones (lóbulo frontal, lóbulo temporal, ganglios basales, cerebelo y tronco cerebral)3,4. Su patogénesis no está esclarecida. Los síntomas y los parámetros hemodinámicos desaparecen completamente cuando se corrige a tiempo la causa subyacente y, aunque los hallazgos de neuroimagen se resuelven de forma más lenta (semanas-meses)5, la neuroimagen de seguimiento con secuencias específicas son necesarias para confirmar el diagnóstico1.
Se presenta un caso de una mujer de 79 años, encontrada caída con desorientación y discurso incoherente, cefalea hemicraneana izquierda de severa intensidad, pulsátil, leve-moderada acompañada de foto/fonofobia, sin náuseas o vómitos, de 7-10 días de evolución. Sin traumatismo craneal, alteración de consciencia, incontinencia esfinteriana o fiebre. Sin antecedentes psiquiátricos o hábitos tóxicos. Antecedentes patológicos: HTA, insuficiencia cardíaca congestiva, úlcera gástrica, sigmoidectomizada hace 6 años (displasia de alto grado) sin quimioterapia previa. Analfabetismo-deterioro cognitivo leve (DCL) hace 2 años, sin evolución. Un año antes la TC craneal mostró ligera leucoaraiosis de predominio frontal. Medicación habitual: omeprazol 20mg, furosemida 40mg, diltiazem 120mg, risperidona 0,5mg. A la admisión: presión arterial 200/78 mmHg y freuencia cardíaca 65. Temperatura axilar 37,4°C. Saturación de O2 98%. ACP: soplo sistólico ii/iii, exploración abdominal normal. Sin edemas en los miembros inferiores. En la exploración neurológica la paciente se encontraba despierta, desorientada T/E, periodos de verborrea, sin agitación. Fondo ojo normal. Paresia facial central izquierda y ligera disartria. Sin déficit motor. Sin signos meníngeos.
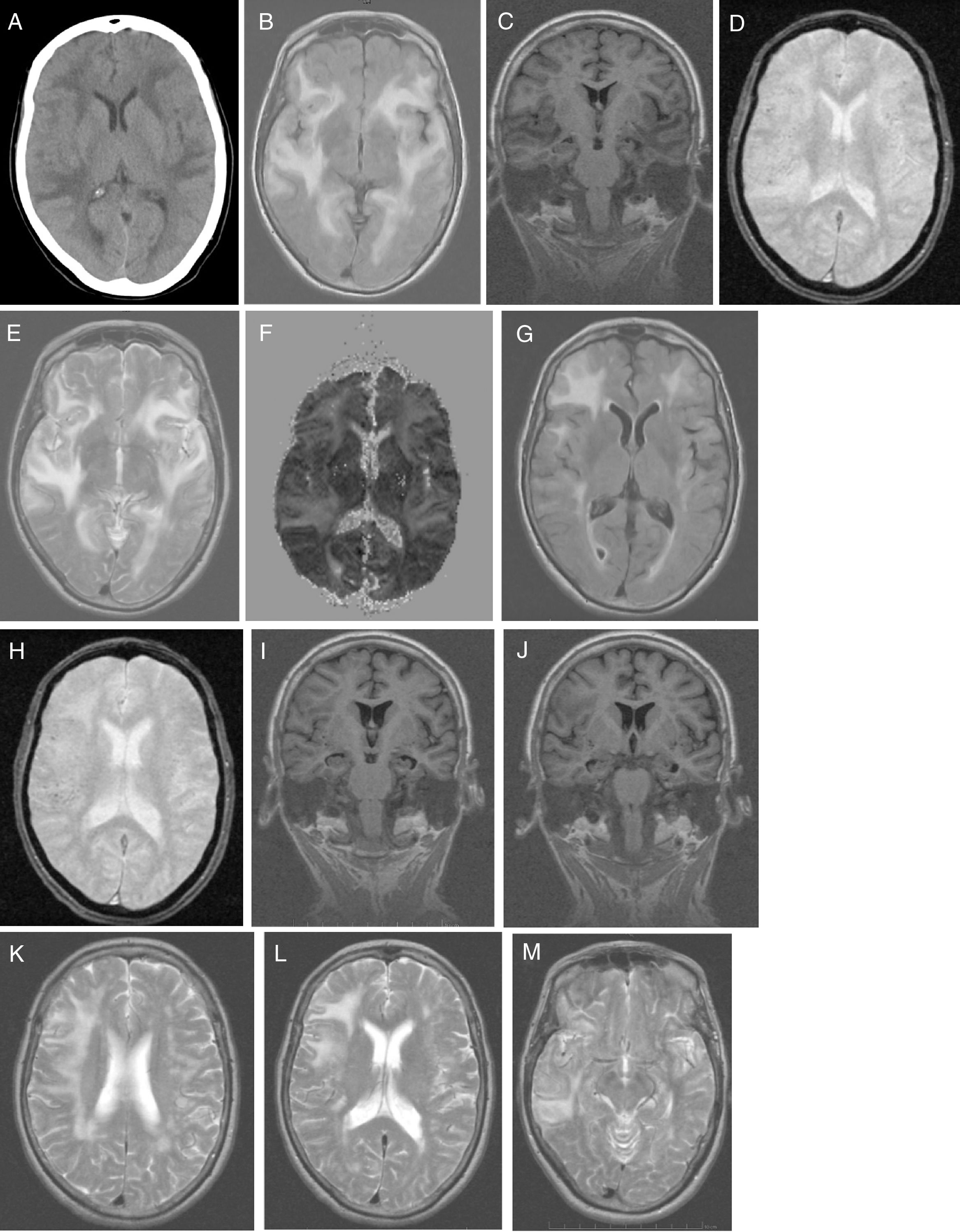
El estudio analítico no mostró alteraciones: hemograma, proteína C reactiva, velocidad de sedimentación globular, coagulación, ionograma, péptido natriurético tipo, función renal, hepática y tiroidea, vitamina B12, ácido fólico, autoinmunidad, anticuerpos anti-Treponema pallidum y virus de la inmunodeficiencia humana 1 y 2. La presencia de tóxicos en orina fue negativa. La radiografía de tórax fue normal. La TC craneal (fig. 1A) identificó extensas áreas hipodensas, bihemisféricas, confluentes y asimétricas, aparentemente edematosas, predominantemente subcorticales, de predominio izquierdo, indicativas de LPR y para caracterizar mejor por RM craneal. Inició tratamiento con captopril por vía oral 25mg cada 8h, furosemida 20 m g iv cada 6h y paracetamol durante 5 días. Hubo mejoría clínica espectacular (sin cefalea, discurso adecuado con la desorientación temporal previamente conocida) y estabilidad hemodinámica. Al 5.° día del inicio de los síntomas la paciente realiza RM craneal (fig. 1B-F), que revela extensa hiperseñal confluente (frontal-temporal-parietal-occipital e insular bilateral) que envuelve la SB (de aspecto digitiforme) y el córtex; escasos focos de hiposeñal yuxtacorticales (microhemorragias). Focos glióticos isquémicos SB en T2 y FLAIR. Sin restricción a la difusión y aumento de señal en mapa de ADC indicativo de LPR; menos común causa tóxico-metabólica. La paciente regresa al domicilio medicada con valsartán 160mg e hidroclorotiazida 12,5mg, con buen control tensional. Fue observada en consulta ambulatoria a las 4 semanas del inicio de los síntomas; el MMSE fue de 14 (orientación 4, fijación 3, atención y cálculo 3, recuerdo diferido 0, lenguaje y construcción 4), se encontraba con discapacidad funcional (AVD básicas conservadas. Las AVD instrumentales, alteradas en las tareas que incluyen cocinar, lavar ropa, uso de transporte, uso de medicación y uso del dinero). Un estudio genético confirmó genotipo APOE ɛ4/ ɛ4. La paciente inició tratamiento con galantamina 8mg de liberación lenta, que no toleró por dolor abdominal; le fue retirada pasando a rivastigmina 4,6mg transdérmica, permaneciendo estable. Al 2.° mes del inicio de los síntomas fueron solicitadas una RM craneal de control (fig. 1G-M) para ver la evolución de la LPR y una PET-PIB cerebral (fig. 2) para excluir enfermedad de Alzheimer (EA). La RM craneal (fig. 1G-M) en T2 reveló hiperseñal menos extensa a nivel fronto-temporal-parietal, sin alteración insular ni occipital. Son visibles microhemorragias a nivel frontotemporal bilateral en T2* y focos glióticos isquémicos en T2. Se aprecia atrofia temporal medial (ATM); estas alteraciones están a favor de la LPR en regresión.
 TC craneal: extensas áreas hipodensas, confluentes y asimétricas, aparentemente edematosas, predominantemente subcorticales, de predominio izquierdo. Patrón holohemisférico bilateral. B-F) RM craneal inicial: corte axial en T1 (B): extensa hiperseñal confluente holohemisférica bilateral de SB y córtex; corte coronal en T1 (C): sin atrofia de hipocampo; corte axial en T2* (D): escasos focos de hiposeñal yuxtacorticales indicativos de microhemorragias cerebrales; corte axial en T2 (E): hiperseñal fronto-opercular, temporoinsular bilateral, a la derecha con extensión parietal y a la izquierda con extensión temporo-occipital medial; mapa de ADC (F): aumento de señal holohemisférica bilateral. G-M) RM craneal de control: corte axial en T1 (G): hiperseñal fronto-temporal bilateral, de menos extensión en el hemisferio izquierdo, sin alteración insular ni occipital; corte axial en T2* (H): microhemorragias yuxtacorticales a nivel fronto-temporal bilateral; cortes coronales en T1 (IJ): atrofia temporal medial y silviana; cortes axiales en T2 (K-M): hiperseñal holohemisférico derecha de menor extensión (K); hiperseñal fronto-parietal derecha (L); hiperseñal temporal derecha (M).")
A) TC craneal: extensas áreas hipodensas, confluentes y asimétricas, aparentemente edematosas, predominantemente subcorticales, de predominio izquierdo. Patrón holohemisférico bilateral. B-F) RM craneal inicial: corte axial en T1 (B): extensa hiperseñal confluente holohemisférica bilateral de SB y córtex; corte coronal en T1 (C): sin atrofia de hipocampo; corte axial en T2* (D): escasos focos de hiposeñal yuxtacorticales indicativos de microhemorragias cerebrales; corte axial en T2 (E): hiperseñal fronto-opercular, temporoinsular bilateral, a la derecha con extensión parietal y a la izquierda con extensión temporo-occipital medial; mapa de ADC (F): aumento de señal holohemisférica bilateral. G-M) RM craneal de control: corte axial en T1 (G): hiperseñal fronto-temporal bilateral, de menos extensión en el hemisferio izquierdo, sin alteración insular ni occipital; corte axial en T2* (H): microhemorragias yuxtacorticales a nivel fronto-temporal bilateral; cortes coronales en T1 (IJ): atrofia temporal medial y silviana; cortes axiales en T2 (K-M): hiperseñal holohemisférico derecha de menor extensión (K); hiperseñal fronto-parietal derecha (L); hiperseñal temporal derecha (M).
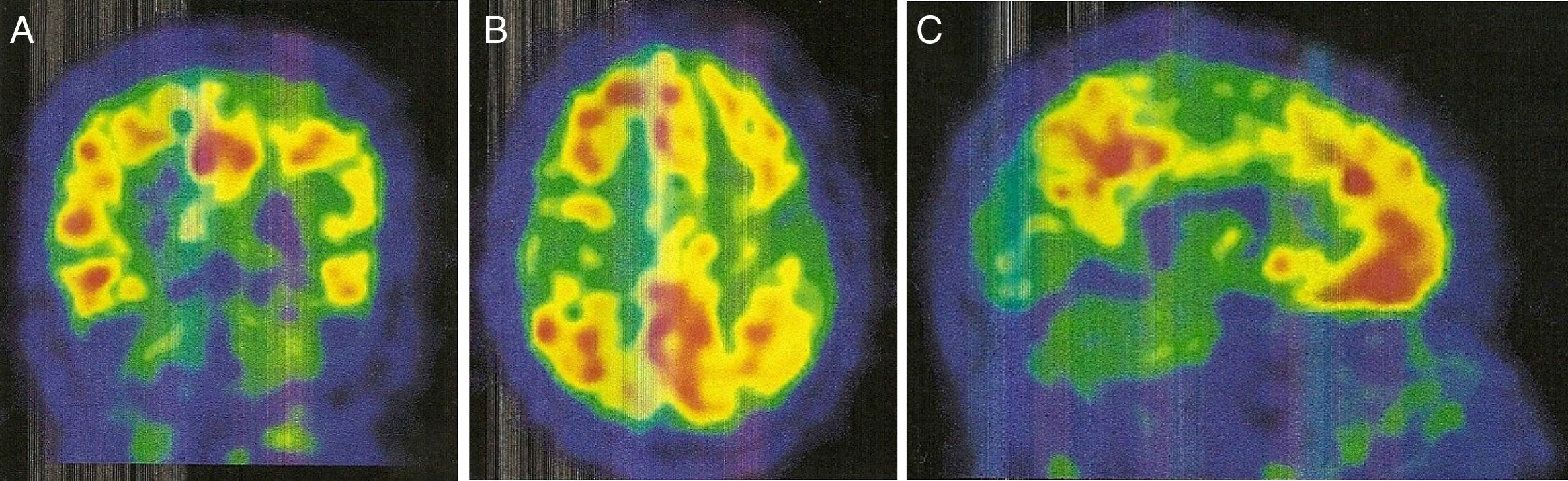
 Corte coronal. B) Corte axial. C) Corte sagital. Aumento de captación de radiofármaco bilateralmente en lóbulo frontal, parietal superior/precuneus, cíngulo posterior y temporal lateral.")
La PET con PIB-C11 (fig. 2) mostró captación aumentada de radiofármaco bilateralmente en lóbulo frontal, parietal superior/precuneus, cíngulo posterior y temporal lateral. Este resultado es compatible con aumento de péptido betaamiloide a nivel cortical.
A los 6 meses del ingreso, la paciente es trasladada a una institución en régimen de hospital de día, permaneciendo estable.
En la evaluación neuropsicológica fueron aplicadas las siguientes escalas: escala de inteligencia de Wechsler para adultos (WAIS-III) y escala de memoria de Wechsler (WMS-III). Fueron identificadas alteraciones de nivel inferior en las siguientes variables: atención, memoria auditiva inmediata, memoria episódica, vocabulario, rapidez psicomotora, memoria de trabajo y operacional, QI verbal y aprendizaje.
En este caso clínico, la edad de presentación de la LPR no es la habitual (próxima a la 8.ª década de la vida). Este patrón extenso de lesión de la SB (holohemisférico bilateral) en paciente con DCL no ha sido descrito en la literatura y no tiene relación con el tipo de situación clínica desencadenante, aunque es más habitual en pacientes trasplantados o con uso de tratamiento inmunosupresor, o en la enfermedad autoinmune6.
En el diagnóstico diferencial de un paciente con LPR y deterioro cognitivo previo debe ser considerada la angiopatía cerebral amiloide (ACA). La leucoencefalopatía reversible asociada a la ACA mostró un patrón típico de predominio posterior7; además, la ACA puede ser un factor de susceptibilidad sobre el cual factores tales como la inflamación o quizás la HTA podían conducir a la progresión de la enfermedad8. Ya han sido descrito casos de demencia rápidamente progresiva acompañada de crisis epilépticas, leucoencefalopatía y efecto de masa, revelando una ACC de evolución fatal, así como de un caso de ACC con leucoencefalopatía reversible y mejoría dramática de la demencia8.
Con relación a la PET-PIB, en la AAC la retención de PIB es intermedia, pero con un incremento relativo de retención occipital comparado con la EA, factor este relacionado con la distribución de material amiloide en la ACA9,10. Por otro lado, la PET-PIB parece tener potencial para predecir hemorragias futuras, así nuevas hemorragias relacionadas con CAA aparecerán preferentemente en sitios de mayor depósito de amiloide9.
Aunque en el caso descrito la progresión a demencia podría ser predecible (paciente anciana con factores de riesgo vascular, analfabetismo-DCL y genotipo APOE ɛ4/ɛ4), lo más probable es que esta LPR particular precipitó la EA; no hubo hemorragia lobar y las microhemorragias cerebrales de localización cortical fueron escasas. Los argumentos a favor del diagnóstico de EA son: deterioro cognitivo progresivo con discapacidad funcional, un marcador topográfico y de degeneración neuronal (ATM), un marcador fisiopatológico (PET-PIB+ con aumento de captación de radiofármaco bilateralmente en áreas típicas de EA) y el hallazgo del genotipo APOE ɛ4/ɛ4 (factor de riesgo de EA).
Sabemos que la presencia de 2alelos ɛ4 es responsable por una disminución en la edad de inicio de presentación de la EA11 y aumenta su incidencia 9,4 veces12. Otro factor importante es que la isomorfa ɛ4 está más frecuentemente asociada con la forma tardía y esporádica de la EA13.
En este caso referido, la presencia del genotipo APOE ɛ4/ɛ4 no afectó a la edad de inicio de la enfermedad.
Ya han sido publicados casos esporádicos de disfunción cognitiva persistente en el contexto de LPR típica14, pero no de EA precipitada por LPR holohemisférica bilateral.
Con relación a la fisiopatología de este cuadro clínico, probablemente esté relacionada con la pérdida de la autorregulación vascular cerebral que origina hiperperfusión y conduce a la rotura de la barrera hematoencefálica con entrada de fluido y productos de la sangre dentro del parénquima cerebral15. Un mecanismo de vasoconstricción cerebral focal por isquemia cerebral es improbable dada la extensión del edema cerebral y no existir signos de isquemia cerebral. La disfunción endotelial ha sido implicada en los casos asociados a preeclampsia, uso de fármacos citotóxicos o enfermedad autoinmune1,2.
La particularidad de este caso clínico es que la paciente, portadora de DCL, progresó a EA después de ser corregida la HTA en contexto de LPR exuberante, cuyo genotipo era APOE ɛ4/ ɛ4. Con este caso, se pretende alertar al clínico sobre el reconocimiento precoz de este síndrome, su pronta intervención terapéutica para evitar secuelas neurológicas y la vigilancia estrecha que requiere todo paciente con LPR a través de una evaluación cognitiva/neuropsicológica a tiempo14. Basándonos en la supuesta reversibilidad de la LPR, podemos subestimar un trastorno cognitivo e impedir el ajuste de nuestros pacientes.
Este caso clínico fue presentado como póster en la LXIX Reunión Anual de la Sociedad Española de Neurología 2017.

