




La enfermedad relacionada con IgG4 (ER IgG4) es una entidad sistémica que incluye a un grupo heterogéneo de condiciones que presentan en común lesiones tumefactas de los órganos afectados, con infiltrado linfoplasmocitario rico en células plasmáticas IgG4 positivas organizados en un patrón estoriforme de fibrosis1. Dado los distintos nombres que recibió esta entidad a lo largo de la historia, la prevalencia e incidencia son desconocidas. Generalmente se presenta entre la 6-7.a década de la vida, afectando de forma predominante a los varones (60-80%)2. El mecanismo fisiopatológico sigue siendo desconocido, aunque las teorías más relevantes postulan un fenómeno autoinmune o alérgico, con una reacción TH-2 desproporcionada a un antígeno aún no identificado3. Las manifestaciones clínicas y formas de presentación son variadas, pudiendo afectar a múltiples órganos, siendo diagnóstico diferencial de distintos procesos neoplásicos, infecciosos e inflamatorios (tabla 1)4. El compromiso del sistema nervioso central (SNC) es infrecuente, aunque existen síndromes neurológicos estrechamente vinculados a ER IgG4, como son la paquimeningitis hipertrófica (PH) y la hipofisitis (HF)5. Se presentan 2 casos de ER IgG4 probable con manifestaciones neurológicas como síntomas iniciales. Nuestro objetivo es analizar las distintas formas de presentación neurológica de la ER IgG4, a fin de aumentar la sospecha diagnóstica del médico neurólogo general, para realizar un tratamiento efectivo precoz.
Criterios diagnósticos clínicos integrales y síndromes reconocidos bajo el espectro de enfermedades relacionadas con IgG4 (ER IgG4)
| Criterios diagnósticos | Síndromes |
|---|---|
| 1 Examen clínico que demuestre masas o agrandamientos difusos/localizados en uno o varios órganos2 Elevadas concentraciones séricas de IgG4 (≥135 mg/dl)3 Examen anatomo-patológico que muestre:a. Marcado infiltrado linfocítico, plasmocitario y fibrosisb. Infiltrado de plasmocitos IgG4+: relación de células IgG4+/IgG+>40% y >10 células plasmáticas IgG4+/campo de alta potencia | -Pancreatitis autoinmune-Síndrome de Mikulicz-Tumor de Küttner-Fibrosis mediastinal-Tiroiditis de Riedel-Fibrosis retroperitoneal-Esclerosis mesentérica-Fibrosis multifocal-Seudotumor inflamatorio-Fibrosis angiocéntrica eosinofílica-Fibroesclerosis multifocal-Periaortitis y periarteritis-Aneurisma aórtico inflamatorio-Nefritis tubulointersticial hipocomplementémica idiopática con depósitos extensos tubulointersticiales-Paquimeningitis hipertrófica-Hipofisitis |
Diagnóstico: definitivo: 1+2+3; probable: 1+3; posible: 1+2.
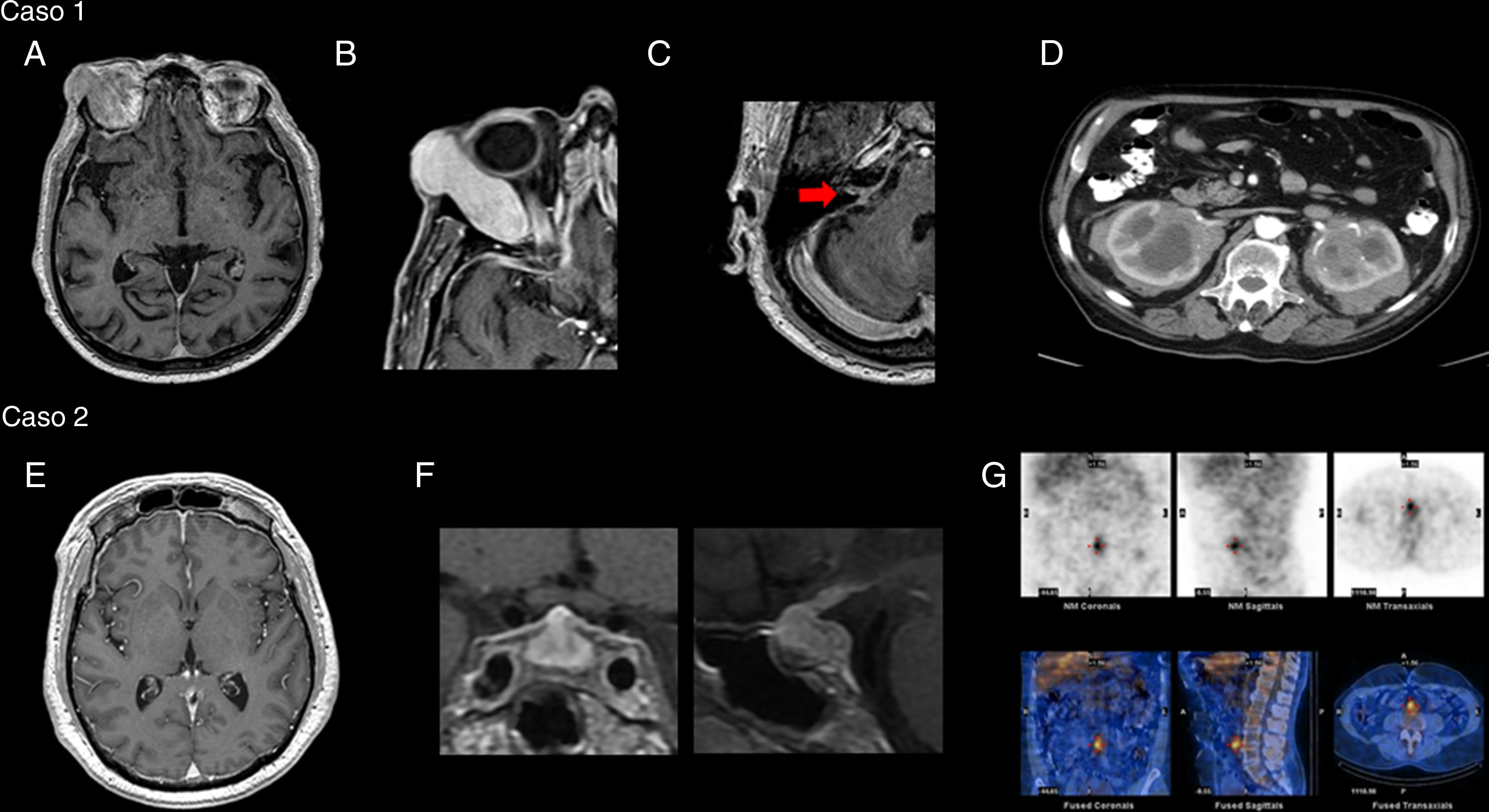
Varón de 77 años de edad, que consultó por diplopía asociada a escalofríos y sudoración nocturna de 6 años de evolución. En el examen físico se objetivó tumoración supraorbitaria derecha con proptosis y limitación de la motilidad ocular ipsilateral, anacusia bilateral y paresia facial periférica derecha. En la resonancia magnética (RM) de cerebro se objetivó refuerzo paquimeníngeo de predominio infratentorial y tumoración que compromete la glándula lagrimal derecha (fig. 1A, B y C). El análisis de laboratorio evidenció eritrosedimentación de 60mm/h y proteína C reactiva de 34,21mg/l. Los valores de IgG total y subtipo 4 en suero fueron normales. En líquido cefalorraquídeo (LCR) presentó hiperproteinorraquia, sin hallazgos microbiológicos ni células neoplásicas. La tomografía computarizada (TC) de abdomen mostró aumento de tamaño de ambos riñones a expensa de lesiones infiltrativas (fig. 1D). El estudio anatomopatológico de la biopsia renal mostró extenso infiltrado linfoplasmocitario e inmunomarcación positiva para CD138 e IgG4. Se interpretó como ER IgG4 probable, y se inició tratamiento con corticoides por vía oral y azatioprina, sin nuevas manifestaciones clínicas, con normalización de los parámetros de inflamación en el laboratorio y resolución del refuerzo paquimeníngeo, tras 6 meses de seguimiento. Sin embargo, se mantuvo la anacusia bilateral como secuela.
 RM de cerebro (T1 con gadolinio): realce paquimeníngeo difuso; B) Lesión expansiva en órbita derecha con refuerzo homogéneo tras la administración de contraste que desplaza al globo ocular; C) Compromiso de paquete facio-acústico (flecha); D) TC de abdomen con contraste: formación expansiva-infiltrativa que compromete ambos riñones, con refuerzo heterogéneo tras la administración de contraste. Caso 2: E) RM de cerebro (T1 con gadolinio): realce paquimeníngeo difuso; F) Incremento difuso del tamaño y señal heterogénea de hipófisis con marcado refuerzo homogéneo tras la administración de contraste; G) Centellograma marcado con galio: hipercaptación peri-aórtica e intercavo-aórtica en topografía del retroperitoneo prevertebral con extensión al sector anterior del espacio discal L4-L5.")
Caso 1: A) RM de cerebro (T1 con gadolinio): realce paquimeníngeo difuso; B) Lesión expansiva en órbita derecha con refuerzo homogéneo tras la administración de contraste que desplaza al globo ocular; C) Compromiso de paquete facio-acústico (flecha); D) TC de abdomen con contraste: formación expansiva-infiltrativa que compromete ambos riñones, con refuerzo heterogéneo tras la administración de contraste. Caso 2: E) RM de cerebro (T1 con gadolinio): realce paquimeníngeo difuso; F) Incremento difuso del tamaño y señal heterogénea de hipófisis con marcado refuerzo homogéneo tras la administración de contraste; G) Centellograma marcado con galio: hipercaptación peri-aórtica e intercavo-aórtica en topografía del retroperitoneo prevertebral con extensión al sector anterior del espacio discal L4-L5.
Varón de 58 años de edad, que consultó por cefalea, visión borrosa y discromatopsia de 4 meses de evolución. En el examen físico se objetivó agudeza visual de 20/70 en ambos ojos. En la RM cerebral se observó aumento difuso del tamaño hipofisario con realce con contraste, asociado a refuerzo paquimeníngeo bifrontal y peritroncal (fig. 1E y F). En el análisis de laboratorio presentó eritrosedimentación de 70mm/h y proteína C reactiva 44,4mg/l asociado a panhipopituitarismo, con valores normales de IgG total y subtipo 4 en suero. En LCR presentó hiperproteinorraquia, sin evidencia microbiológica ni neoplásica. La TC de abdomen y centellograma con galio detectó imagen compatible con fibrosis retroperitoneal (fig. 1G). El estudio anatomopatológico de biopsia de hipófisis mostró extenso infiltrado linfoplasmocitario e inmunomarcación positiva para CD138 e IgG4. Se interpretó como ER IgG4 probable y debido al compromiso severo de ambos nervios ópticos se comenzó tratamiento combinado con corticoides y ciclofosfamida endovenosos, asociado a plasmaféresis con respuesta regular inicialmente. Posteriormente, continuó tratamiento con azatioprina evolucionando a los 20 meses sin nuevas manifestaciones clínicas, sin secuelas visuales, con normalización de los parámetros de inflamación y reducción del tamaño hipofisario. El panhipopituitarismo secuelar se trató con sustitución hormonal.
Las formas de afectación del SNC de presentación más típicas de la ER IgG4 son la PH y la HF, aunque el seudotumor orbitario y la fibrosis de la fosa pterigopalatina también pueden generar compromiso neurológico compresivo indirecto4. La PH se manifiesta frecuentemente con cefalea y neuropatía craneana, reflejando la compresión de diversas estructuras del sistema nervioso o de la vasculatura que lo irriga. Los hallazgos en RM pueden evidenciar compromiso paquimeníngeo difuso y/o focalizado en forma de masa6. La HF se manifiesta con cefalea y trastornos visuales, asociado a síntomas generados por disfunción hormonal. En la RM puede observarse engrosamiento difuso o la formación de masas en la hipófisis y el tallo. Ambas entidades pueden presentarse de forma concomitante7. El compromiso del SNC de la ER IgG4 representa un diagnóstico diferencial de diversos procesos: infecciosos (neurosífilis, tuberculosis, micosis sistémicas), inflamatorios (granulomatosis de Wegener, arteritis de células gigantes, artritis reumatoidea y neurosarcoidosis), neoplásicos (carcinomatosis meníngea, linfomas, meningiomas, craneofaringiomas, adenomas o germinomas de hipófisis), hasta incluso procesos locales benignos (rotura de quiste de hendidura de Rathke)4,6,7.
Se dispone de múltiples criterios diagnósticos que son útiles en la práctica clínica diaria. Sin embargo, no se encuentran internacionalmente estandarizados en la actualidad. Los más utilizados son los que combinan criterios clínicos, de laboratorio y anatomopatológicos (tabla 1)8,9, siendo necesario la presencia de los 3 parámetros para considerar a la ER IgG4 como definitiva. No obstante, hasta el 50% de los pacientes con diagnóstico confirmado por biopsia pueden tener valores normales de IgG4 en suero10, por lo que la presencia de valores normales no debe condicionar la conducta terapéutica, como ocurrió en nuestros 2 casos. Los pacientes que muestran mayor actividad de la enfermedad tienen recuentos de plasmoblastos circulantes elevados detectados por citometría de flujo, incluso con valores de IgG4 sérico normal. Sin embargo, aún no hay trabajos que validen su utilidad diagnóstica11 y el gold standard continúa siendo el estudio anatomopatológico del órgano afectado12. Actualmente, no existen esquemas terapéuticos estandarizados. Sin embargo, el consenso es iniciar tratamiento inmunosupresor en pacientes con enfermedad sintomática activa y aquellos con manifestaciones subclínicas que pudieran conducir potencialmente a secuelas graves e irreversibles13. Los glucocorticoides constituyen el agente de primera línea como terapia de inducción, pudiendo iniciarse por vía oral o incluso endovenosa, en aquellos casos con riesgo funcional inminente, como el caso de nuestro paciente con déficit visual severo. Otros agentes inmunosupresores (azatioprina, micofenolato, 6-mercaptopurina, metotrexato, tacrolimus, ciclofosfamida) se han utilizado como agentes-ahorradores de corticoides e incluso como monoterapia cuando existen contraindicaciones para los corticoides, aunque la eficacia de los mismos no ha sido probada en estudios prospectivos13. Al igual que en otras enfermedades autoinmunes mediadas por anticuerpos IgG414, el rituximab demostró ser efectivo en ER IgG4 en estudios retrospectivos, incluso en pacientes que han fallado con agentes-ahorradores de esteroides13. Luego del tratamiento de inducción satisfactorio, algunos pacientes se benefician con el mantenimiento de inmunosupresores (dosis bajas de corticoides, agentes-ahorradores de esteroides o rituximab) aunque no se ha determinado la duración óptima del mismo y dependerá de cada caso en particular13.
En conclusión, si bien la ER IgG4 es una enfermedad sistémica inflamatoria con compromiso de múltiples órganos, puede comenzar únicamente con síntomas neurológicos. Debe sospecharse en pacientes con PH e HF, en los cuáles se han descartados otras causas más frecuentes. Un valor normal de IgG4 en suero no debe excluir el diagnóstico ni condicionar la conducta terapéutica. El estudio anatomopatológico del órgano afectado sigue siendo el pilar más importante en el diagnóstico. Es de vital consideración el diagnóstico y tratamiento precoz, ya que una vez consolidada la fibrosis de los tejidos, las consecuencias suelen ser irreversibles. Los corticoides son de elección, debiendo considerarse al rituximab en aquellos casos severos o refractarios.

