



Deletions in chromosomal region 17p13.3, which includes the PAFAH1B1 gene (also known as LIS1), are associated with cerebral malformations typical of lissencephaly, such as absence of gyri and cortical thinning. Phenotypes vary greatly, ranging from isolated lissencephaly to Miller-Dieker syndrome (MDS; OMIM #247200).1 Miller first described MDS in 1963, as a genetic disorder with variable clinical expression. The condition may be caused by a wide range of genetic alterations, including deletions, duplications, contiguous gene syndromes, or mutations affecting PAFAH1B1.2 This explains the frequent combination of different phenotypes: we may find 17p13.3 microdeletions with no lissencephaly, indicating that PAFAH1B1 expression is normal.3
We describe the case of an 11-year-old girl from Colombia, born to non-consanguineous parents. Her mother was a 27-year-old, gravida 4, para 2 woman who had experienced 2 miscarriages following ectopic pregnancies. The patient was the mother's fourth pregnancy, which was complicated by placenta praevia, gestational diabetes, and Rh incompatibility. The patient was delivered vaginally at 37 weeks of pregnancy, with a birth weight of 2500g, a length of 36cm, and clinical signs of intrauterine growth restriction. She underwent surgical closure of patent ductus arteriosus after birth; during development, she displayed psychomotor retardation, language impairment, and short stature.
The physical examination revealed a height of 121cm (3rd percentile, −2 SD), a weight of 28.3kg (3rd percentile), a head circumference of 54cm (50th-75th percentile), high anterior hairline, wide forehead, telecanthus, smooth philtrum, micrognathia, wide palate, short neck, clavicle hypoplasia, mild pectus excavatum, long thorax, pelvic tilt to the left, bilateral clinodactyly, cutaneous syndactyly affecting the second and third phalanges, hyperextension of the elbow (200°), and a hand length of 14cm (below the 5th percentile).
Complementary testing (echocardiography; urinary tract, renal, and abdominal ultrasound; bone age study; glucose and thyroid-stimulating hormone levels) yielded normal results. G-banding karyotyping at a resolution of 650 bands showed normal chromosome complement (46 chromosomes, XX).
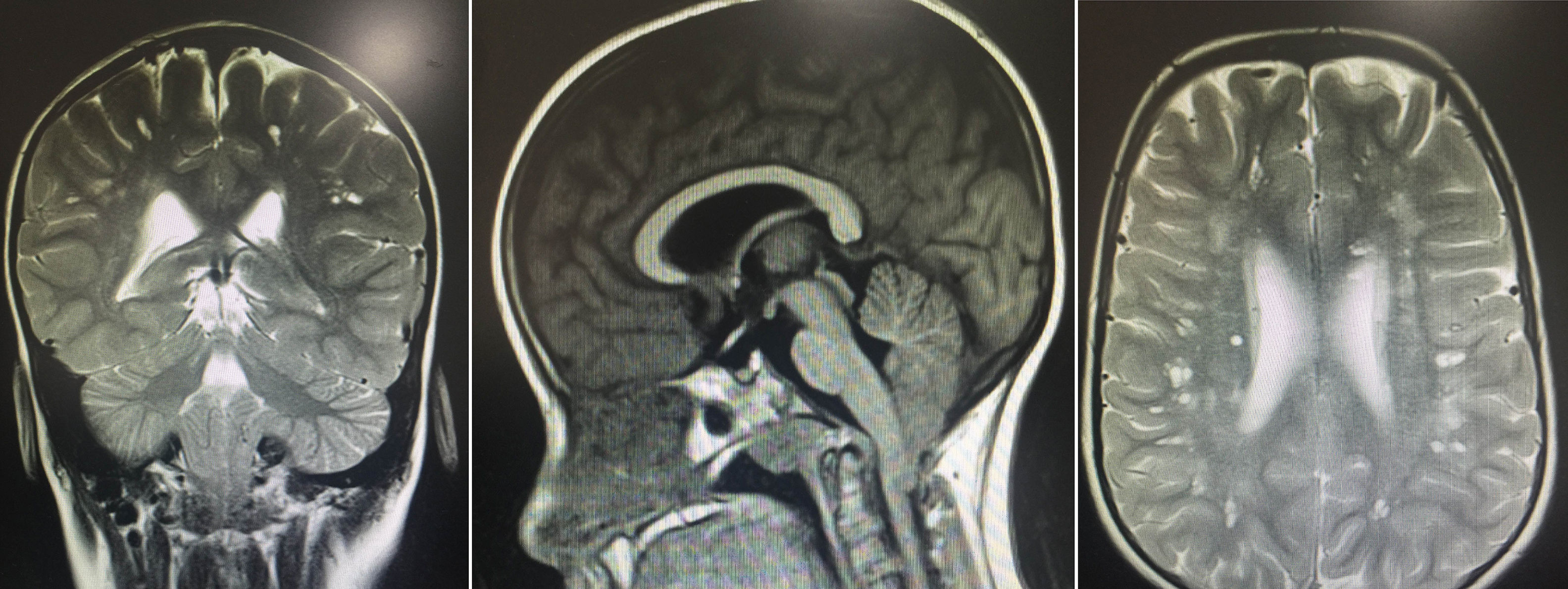
The results of the neuropsychological evaluation revealed poor intellectual function: a verbal IQ score of 49, a performance IQ score of 47, and a full-scale IQ score of 41 on the Wechsler Intelligence Scale for Children. A brain MRI revealed multiple lesions, especially in the subcortical white matter bilaterally, resembling small spaces with CSF-like behaviour. Images also showed hyperintense areas, some surrounding the lesions, giving the brain a spongiform appearance, with no signs of lissencephaly. We also observed downward displacement of the cerebellar tonsils through the foramen magnum (type I Chiari malformation) (Fig. 1).
.")
Brain MR images showing multiple lesions, especially in the subcortical white matter bilaterally, resembling small spaces with CSF-like behaviour in all sequences, and hyperintense areas, some surrounding the lesions, giving the brain a spongiform appearance. The images also show moderate downward displacement of the cerebellar tonsils through the foramen magnum (type I Chiari malformation with no morphological changes).
The neurological and physical findings led us to suspect a genetic disorder. Our first diagnostic hypothesis was a microdeletion syndrome; microarray-based comparative genomic hybridisation detected a 2.19-MB deletion in 17p13.3, (525-2 190 945)x1, encompassing TUSC5, YWHAE, CRK, MYO1C, and SKIP, but not PAFAH1B1.
Chromosome region 17p13.3 is known to be unstable as it contains extensive repetitive sequences. PAFAH1B1 (coded as LIS1) haploinsufficiency causes isolated lissencephaly or MDS, depending on the size of the deletion. Microdeletion mapping has recently identified MDS in the MDS telomeric critical region, which is associated with different overlapping phenotypes.4
Distal 17p13.3 microdeletion syndrome not encompassing PAFAH1B1 is a relatively new microdeletion syndrome, previously reported in 16 patients.4 Patients may present terminal or interstitial deletions of varying size, with haploinsufficiency involving several genes, including YWHAE, TUSC5, CRK, and MYO1C.2
The syndrome is characterised by facial dysmorphism, mainly in the form of wide forehead, broad nasal base, and prominent lips.5 Imaging studies usually show leukoencephalopathy, white matter abnormalities, and white matter structural alterations; deletion of YWHAE is therefore thought to affect white matter and myelination. This conclusion is based on the fact that simultaneous deletion of YWHAE, CRK, and PAFAH1B1 results in severe lissencephaly.6
Our patient displayed psychomotor retardation, cognitive deficits, facial dysmorphism, neuropathy of the lower limbs, and multiple white matter abnormalities; this is consistent with the results of other series, which also report no muscle tone alterations in patients with PAFAH1B1 deletion. This adds to the hypothesis that YWHAE and CRK are involved in myelination. Type I Chiari malformation was found in 3 patients of a series of 12.4 This type of malformation has been associated with numerous congenital syndromes and may be caused by reduced posterior fossa size, resulting in migration of its neuronal contents, probably in association with alterations in CSF homeostasis, as in the case presented here.7 All individuals with chromosome 17p13.3 microdeletions have learning difficulties.4
According to Cardoso et al.,8 other genes besides PAFAH1B1 are involved in determining MDS phenotype. This supports the hypothesis that MDS is a contiguous gene syndrome; patients with 17p13.3 deletion not encompassing PAFAH1B1 may present similar dysmorphic facial features. We found a genotypic correlation between our patient and patients from other series, where the most frequently involved genes are SKIP, MYO1C, and CRK. In most published cases, the critical region spans 258kb (Chr17: 1136270–1394633) and includes 6 genes (exons 2-3 of TUSC5, YWHAE, CRK, MYO1C, SKIP, and exons 1-4 of PITPNA) that play a crucial role in the CNS9,10; these genes were also affected in our patient.
YWHAE is the candidate gene for the dysmorphic phenotype associated with MDS.11 The gene encodes 14-3-3ɛ protein, which plays a critical role in neuronal migration. This protein binds to phosphorylated Cdk5/p35, sustaining phosphorylation; in this state, the protein constitutes a cytoplasmic regulatory target for neuronal migration. Therefore, impairment of this protein results in neuronal migration defects and such typical features as Arnold-Chiari malformation and white matter lesions.12,13
Other genes affected include CRK (v-crk sarcoma virus CT10 oncogene homologue) and RPA, which are located in the region affected by the deletion. These genes regulate growth through interaction with the insulin-like growth factor 1. CRK also plays a role in cell differentiation and has been found to be involved in mitogenesis, neuronal migration, neural crest cell migration, and craniofacial development.14
Postnatal growth retardation has been attributed to CRK deletion,5 which probably causes growth restriction, as previously reported by Nagamani et al.5 and Mignon-Ravix et al.15; this is consistent with our case. TUSC5 plays a role in facial dysmorphism5; patients with chromosome 17p13.3 microdeletion show the same dysmorphic facial features as those associated with MDS despite LIS1 being unaffected.
Our patient receives mainly symptomatic treatment and undergoes frequent neurology, endocrinology, and genetic evaluations. She received growth hormone treatment for short stature, growing 12.5cm in 2.2 years (from −5 SD to −3 SD). Growth hormone treatment was discontinued after diagnosis of Arnold-Chiari malformation. She also receives speech, occupational, and neurodevelopmental therapy.
No large series have been published due to the low incidence of the disease. With a view to providing a more accurate prognosis, long-term follow-up of these patients is necessary to determine whether early findings of leucoencephalopathy may constitute a sign of leucodystrophy, and to ascertain whether these patients usually present other endocrine disorders, as in the case presented here. The significance of our results is therefore uncertain.
FundingThis study was funded by Universidad Icesi and the Centre for Research into Congenital Anomalies and Rare Diseases (CIACER).
Author contributionsAll authors revised the manuscript and approve publication of the article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Candelo E, Caicedo G, Mejia L, Pachajoa H. Síndrome de microdeleción 17P13.3 sin afectación del gen PAFAH1B1. Neurología. 2019;34:482–484.
