



La deleción del 17p13.3, incluyendo el gen PAFAH1B1 o LIS1, se asocia a malformaciones cerebrales tipo lisencefalia; caracterizada por reducción de giros y adelgazamiento cortical, sin embargo existen fenotipos variables que van desde lisencefalia aislada hasta síndrome de Miller-Dieker (SMD) (MM: 247200)1. Descrito por primera vez por Miller en 1963 como una condición genética con una expresión clínica variable, el cual puede ser causado por diferentes anormalidades genéticas tales como: deleción, duplicación, síndrome de genes contiguos y mutación del gen PAFAH1B12. Por tal razón la combinación de fenotipos es frecuente y es posible encontrar microdeleciones de la región 17p13.3 en ausencia de lisencefalia implicando que la expresión del gen PAFAH1B1 es normal3.
Describimos una paciente de origen colombiano, sexo femenino de 11 años de edad al momento de la evaluación, producto de unión entre padres no consanguíneos. Madre de 27 años, grávida cuatro, dos partos, dos abortos ectópicos, producto de la cuarta gestación, la cual cursó con placenta previa, diabetes gestacional e incompatibilidad del Rh. Gestación de 37 semanas, parto vaginal, peso al nacer de 2.500g, talla de 36cm y signos clínicos de restricción de crecimiento intrauterino. Al nacimiento presentó ductus arterioso persistente con corrección quirúrgica, y durante el desarrollo retraso psicomotor, trastorno del lenguaje y talla baja.
Observándose al examen físico: talla de 121cm en percentil -3-2 DS, peso de 28,3kg en percentil 3 DS y perímetro cefálico 54cm en percentil 50-75; implantación del cabello anterior y alta, frente amplia, telecanto, filtrum plano, micrognatia con paladar ancho, cuello corto, hipoplasia clavicular, ligero pectun excavatum, tórax largo, basculación pélvica izquierda, clinodactilia bilateral, sindáctila cutánea en segundo y tercer artejo, hiperextensión de 200° del codo y, longitud de la mano de 14cm por debajo del percentil 5.
Con estudios de extensión como: ecocardiograma, ecografía de vías urinarias, renal, abdomen, carpograma, glucemia, hormona estimulante de la tiroides (TSH) todos en parámetros normales. Cariotipo con bandeo G con resolución de 650 bandas mostrando un complemento cromosómico reportado como normal (46, XX).
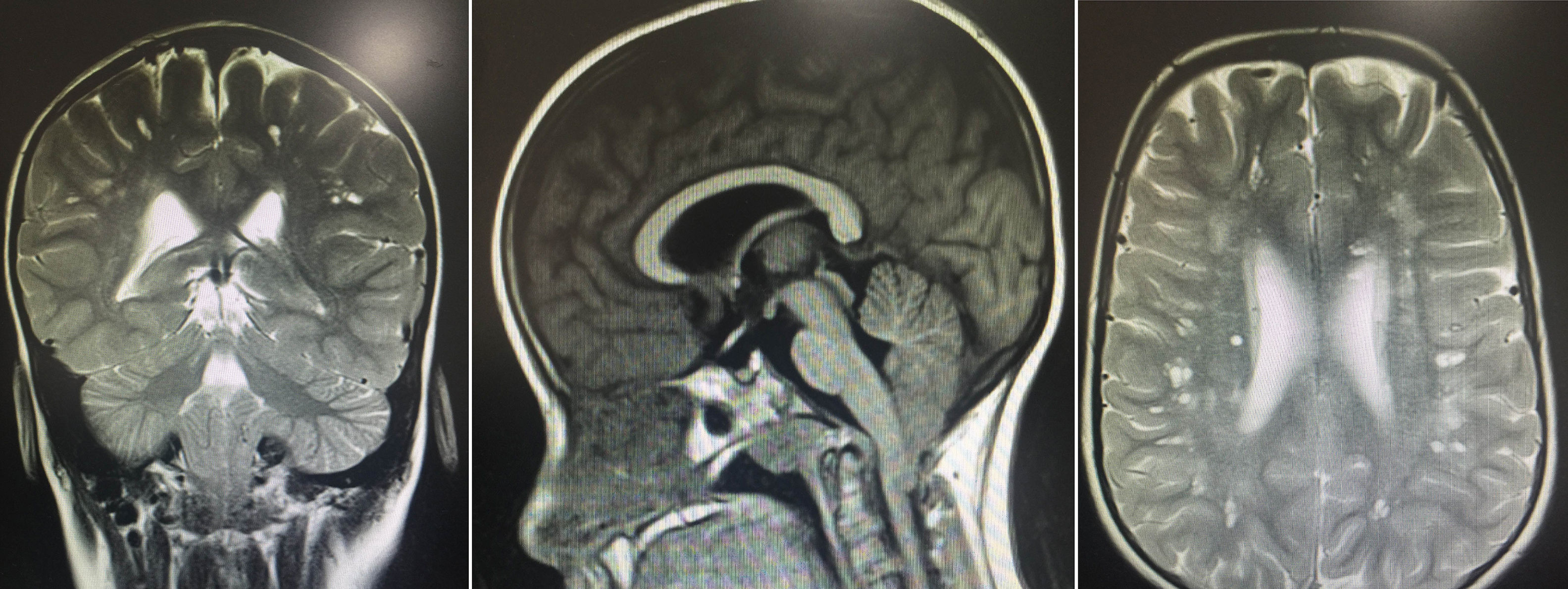
Se encontraron resultados anormales en test neuropsicológico: deficiente coeficiente intelectual verbal de 49, manipulativa de 47, total de 41 en la escala de Wechsler. Se realizaron imágenes diagnósticas, resonancia magnética nuclear con múltiples lesiones especialmente localizadas en la sustancia blanca subcortical en forma bilateral, caracterizado por pequeños huecos de comportamiento similar al líquido cefalorraquídeo, en todas las secuencias áreas de hiperintensidad, algunas de las cuales rodean estas lesiones y dan un aspecto espongiforme, sin signos de licencefalia. Adicionalmente moderado descenso de las amígdalas cerebelosas a través del agujero magno, configurando una enfermedad de Chiari tipo I (fig. 1).

Se observan múltiples lesiones especialmente localizadas en la sustancia blanca subcortical en forma bilateral, caracterizadas por pequeños huecos de comportamientos en la língula similar al del líquido cefalorraquídeo en todas las secuencias y áreas de hiperintensidad de señal, algunas de las cuales rodean estas lesiones y dan un aspecto espongiforme. Moderado descenso de las amígdalas cerebelosas a través del agujero magno configurando una enfermedad de Chiari I sin cambios morfológicos.
Se concluye que la sumatoria de hallazgos neurológicos y al examen físico sería explicado por otra patología, y que tal fenotipo podría tener un origen probablemente genético. Se estableció como primera posibilidad diagnóstica un síndrome por microdeleción por lo que se solicitó prueba de hibridación genómica comparativa con microarreglos, la cual concluye una deleción 17p13.3 de un tamaño de 2,19MB, (525-2,190,945)x1. No afectando el gen PAFAH1B1, abarcando la zona crítica TUSC5, YWHAE, CRK, MYO1C, SKIP.
El cromosoma 17p13.3 contiene extensivas secuencias repetitivas, por lo tanto es reconocido como una región inestable. La haploinsuficiencia del PAFAH1B (codificado como LIS1) causa lisencefalia aislada o SMD, dependiendo del tamaño de la deleción afectado. Recientemente con el mapeo de las microdeleciones se han identificado SMD en la región crítica telomérica, asociado con la superposición de los diferentes fenotipos4.
La deleción distal del 17p13.3 sin involucrar el PAFAH1B1 es un síndrome emergente de microdeleción reportado anteriormente en 16 pacientes4. Los pacientes pueden tener deleción terminal o intersticial de tamaño variado, con haploinsuficiencia de genes que comprenden los siguientes; YWHAE, TUC5, CRK y MYO1C entre otros2.
Este síndrome se caracteriza por dimorfismo facial, que incluye principalmente: frente amplia, base nasal amplia, labios prominentes5. En los hallazgos de imagen lógicos es común encontrar leucoencefalopatía y presencia de anormalidades en la sustancia blanca, cambios que afectan la distribución de la sustancia blanca, por lo que se considera que la presencia de la deleción de YWHAE y CRK podría afectar la sustancia blanca y la mielinización. Este hallazgo se concluye debido a que la deleción sinérgica del YWHAE y CRK sumado al PAFAH1B1 resulta en el fenotipo de licencefalia más severa6.
En el caso presentado se encuentra paciente con retraso del desarrollo psicomotor y déficit cognitivo, dimorfismo facial, neuropatía de miembros inferiores y múltiples anormalidades de la sustancia blanca, congruente con lo reportado en otras series donde muestran que los pacientes con deleción del PAFAH1B1 no presentan anormalidades en el tono muscular, por lo que este hallazgo refuerza la teoría de que los genes YWHAE y CRK participan en el proceso de mielinización. En una serie de 12 pacientes se reportó la presencia de malformación Chiari tipo I en 3 pacientes4, esta malformación se ha asociado a múltiples síndromes congénitos y podría resultar de la alteración en la migración neuronal entre la fosa posterior y el contenido neuronal, posiblemente asociado con anormalidades en la regulación de la homeostasis del fluido cerebroespinal, condición que se encontraba presente en el caso reportado7. Todos los individuos con 17p13.3 desarrollan retraso variable en el aprendizaje4.
Los resultado de la base de datos de Cardoso et al.8 muestran que existen otros genes involucrados en generación del fenotipo de los pacientes con SMD diferentes al gen PAFAH1B1, soportando el concepto de que el SMD es un síndrome de deleción contigua y en pacientes con deleción 17p13.3 sin afección del PAFAH1B1 presentan el mismo espectro de dimorfismo facial. Adicionalmente, se encuentra una correlación genotípica entre los pacientes reportados en otras series y la paciente presentada, donde la mayoría de genes involucrados fueron: SKIP, MYO1C y CRK. En la mayoría de los casos reportados en la literatura la región crítica abarca 258kb (Chr17: 1136270-1394633) e incluye seis genes (exones 2-3 de TUSC5, YWHAE, CRK, MYO1C, SKIP y exones 1-4 de PITPNA) con función central en el sistema nervioso central9,10 los cuales estuvieron involucrados en nuestro paciente.
YWHAE es el gen candidato de generar el fenotipo dismórfico asociado al SMD11, adicionalmente este gen codifica la proteína 14-3-3ɛ, la cual tiene un papel crucial en la migración neuronal. Esta proteína se une a la CDK5/p35 fosforilada y la mantiene en su estado fosforilado, en este estado, la proteína es una diana reguladora citoplasmática de la migración neuronal. Por lo tanto, cuando se afecta genera un defecto en la migración neuronal y genera características típicas del síndrome tales como; malformación de Arnold Chiari y lesiones en la sustancia blanca12,13.
Entre otros genes afectados encontramos CRK (v-CRK sarcoma virus CT10 oncogén homólogo) y RPA, se encuentran en la región donde ocurre la deleción. Estos genes se encargan de regular el crecimiento, produciendo un efecto en el factor de crecimiento relacionado con la insulina 1. CRK tiene un papel adicional en la diferenciación celular y se ha encontrado involucrado en la mitogénesis, migración neuronal y de la cresta neural y desarrollo craneofacial14.
El retardo en el crecimiento posnatal de estos pacientes se ha atribuido a la concurrente deleción del CRK5, probablemente genera la restricción en el crecimiento, descrito previamente por Sreenath Nagamani et al. y Mignon-Ravix et al.15, hallazgo congruente con el caso clínico presentado, el TUSC5 tiene un rol en el dimorfismo facial5, por lo tanto pacientes con microdeleción 17p13.3 presentan las características dimorfofaciales de los pacientes con SMD a pesar de no tener afectación del gen LIS1.
El momento del manejo de la paciente es principalmente sintomático y expectante con controles frecuentes con neurología, endocrinología y genética. Se ha realizado manejo con hormona de crecimiento para la talla baja, con crecimiento de 12,5cm en un periodo de 2,2 años, pasando de -5 desviaciones estándar a -3 desviaciones estándar. Recibió manejo con hormona de crecimiento, el cual fue suspendido por el hallazgo posterior del Arnold Chiari. Adicionalmente se realizan terapias de lenguaje, ocupacional y de neurodesarrollo.
Debido a la ausencia de series largas por la baja frecuencia de esta enfermedad y para una mayor certeza del pronóstico de los pacientes, es necesario el seguimiento a largo plazo de dichos pacientes para determinar si los hallazgos tempranos de leucoencefalopatía podrían ser un signo de leucodistrofia, y vigilar si en la mayoría de estos pacientes hay presencia de otras enfermedades endocrinas como las que presentó nuestra paciente, por esto la significación de estos resultados continúa siendo incierta.
FinanciaciónLa realización de este trabajo ha sido financiada por la Universidad Icesi y el Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER).
Autoría/colaboradoresEste manuscrito ha sido revisado por todos los autores citados y avalado para su publicación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

