



The histone acetyltransferase KAT6B is a component of the MOZ/MORF protein complex of epigenetic readers. The protein participates in both transcriptional activation and repression, and is involved in the development of the cerebral cortex.1,2 As with other genes responsible for chromatin regulation, KAT6B dysfunction causes a multisystem developmental disorder.1
Some of the disorders known to be linked to the KAT6B gene include genitopatellar syndrome and Say-Barber-Biesecker-Young-Simpson syndrome (SBBYSS), also known as the Say-Barber-Biesecker-Young-Simpson variant of Ohdo syndrome.1,3–8
We describe the case of a boy with a de novo KAT6B mutation presenting a phenotype compatible with SBBYSS, without intellectual disability but with specific language impairment and severe attentional disorder.
The patient was an 8.5-year-old boy, an only child born to healthy parents of Spanish origin. The patient was brought to our centre due to severe learning difficulties, language problems, and attentional disorder; he did not present restricted interests or stereotypies, and displayed marked social interest. The patient displayed poor vocabulary, and made significant grammatical errors. He had no relevant family history. He was born after a normal pregnancy and delivery, with a birth weight of 2970 g (10th percentile) and a birth length of 49 cm (25th percentile). The patient had undergone surgery due to hypospadias and cryptorchidism. He presented severe language delay, using very few words at the age of 3 years. Motor development was normal, and the patient had started walking at 14 months of age. At the age of 6 years, a neuropsychological evaluation (WPPSI-III) revealed a verbal intelligence quotient (IQ) of 58 (percentile 0.3) (comprehension subtest, with a typical score of 1), and a manipulative IQ of 93 (33rd percentile); total IQ could not be calculated due to the difference between scales.
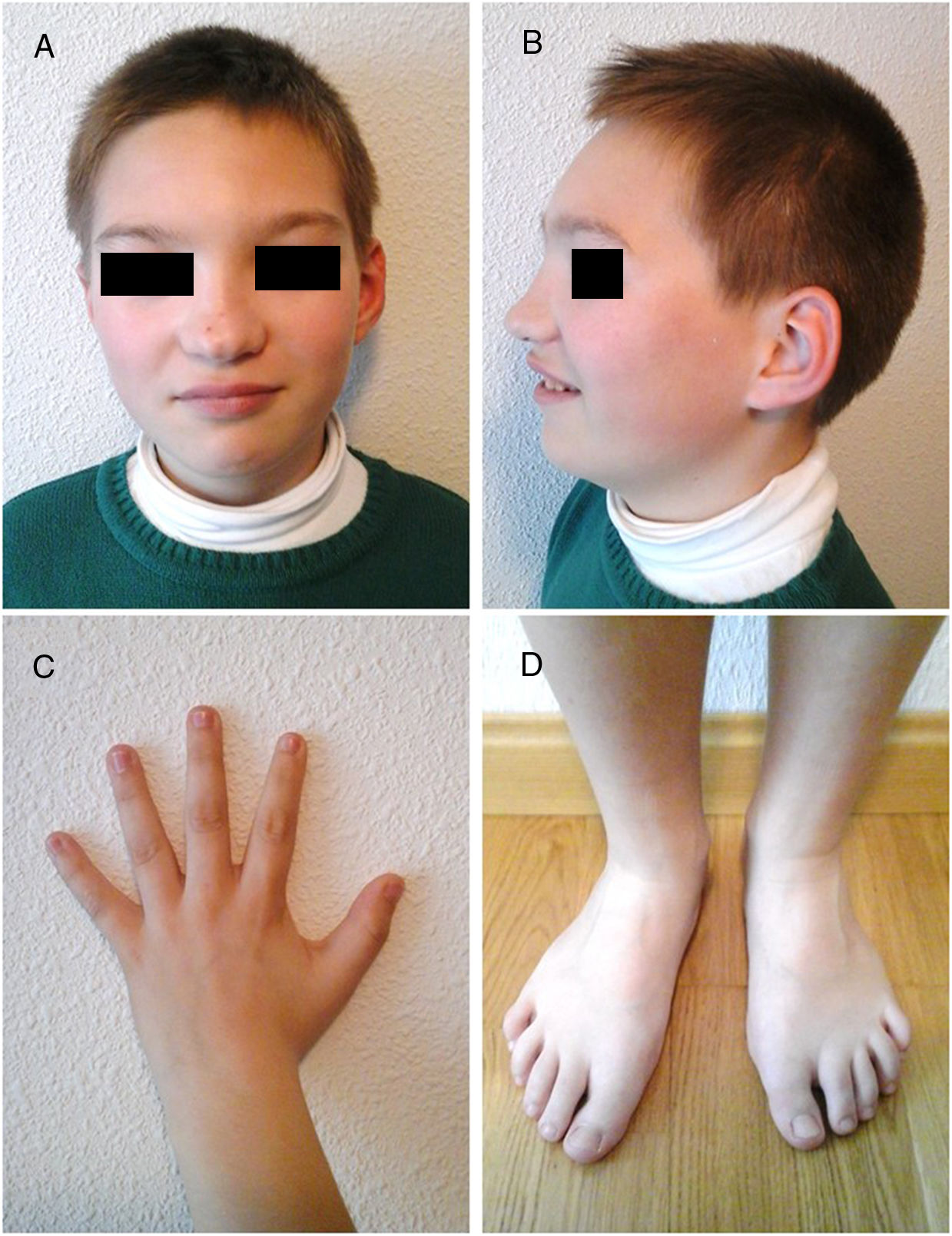
The physical examination detected no focal neurological signs, although the patient displayed severe language impairment (poor vocabulary and comprehension). He weighed 38 kg and was 140 cm tall (97th percentile). He also presented several dysmorphic features: blepharophimosis, ptosis, hypertelorism, bulbous nose, mild retrognathia, limited ability to separate the fingers in both hands, mild limitations on complete extension of the knees, and long toes on both feet (Fig. 1).
 Facial features: blepharophimosis, ptosis, hypertelorism, bulbous nose, and mild retrognathia. C) Limitations in thumb abduction. D) Long toes in both feet.")
Array testing and sequencing of the PTEN and NSD1 genes yielded normal findings. A brain MRI scan showed no abnormalities.
A neuropsychological evaluation revealed IQ scores of 108 and 102 in the TONI-2 and Leiter-R scales, respectively. Language assessment (ITPA, Peabody, and BLOC-R) revealed a psycholinguistic age of 6 years. The continuous performance test revealed severe attentional disorder (omissions and Hit reaction time variability above the 95th percentile).
At the age of 10 years, the patient and his parents underwent exome sequencing, which identified a de novo nonsense mutation in exon 16 of the KAT6B gene (hg19; chr 10: 76781739; NM_012330.3; c.3122C > A, p.Ser1041*). The mutation, which was subsequently confirmed by Sanger sequencing, was not listed on any genetic database and had not previously been reported in the literature.
Subsequent cardiac and thyroid status examinations revealed no significant alterations.
Our patient’s clinical phenotype is similar to those previously described in the literature. SBBYSS is a rare syndrome characterised by blepharophimosis and intellectual disability.3–8 All the cases reported in the literature to date were associated with severe cognitive disorders. Patients with SBBYSS frequently present other abnormal features or malformations, including hypomimia, bulbous nose, bone and ligament alterations, and long toes.3
Mutations in the KAT6B gene have been linked to genitopatellar syndrome and SBBYSS. These syndromes probably lie on a continuum of developmental disorders, with mutations located in the last exons being associated with clinical profiles more similar to that of SBBYSS.5,9
Ours is the first reported case of a patient with SBBYSS without intellectual disability, and presenting a novel mutation. This case suggests an association between neurodevelopmental disorders and mutations in genes classically linked to intellectual disability and autism.10 Genetic studies should be considered in the diagnosis of patients with severe language impairment.10
FundingThe authors have received no funding for this study.
Please cite this article as: Fernández-Mayoralas DM, Calleja-Pérez B, Álvarez S, Fernández-Jaén A. Mutación de novo en KAT6B, síndrome Say-Barber-Biesecker-Young-Simpson y trastorno específico del lenguaje. Neurología. 2020;35:601–603.
