



Ataxia and hereditary spastic paraplegia are rare neurodegenerative syndromes. We aimed to determine the prevalence of these disorders in Spain in 2019.
Patients and methodsWe conducted a cross-sectional, multicentre, retrospective, descriptive study of patients with ataxia and hereditary spastic paraplegia in Spain between March 2018 and December 2019.
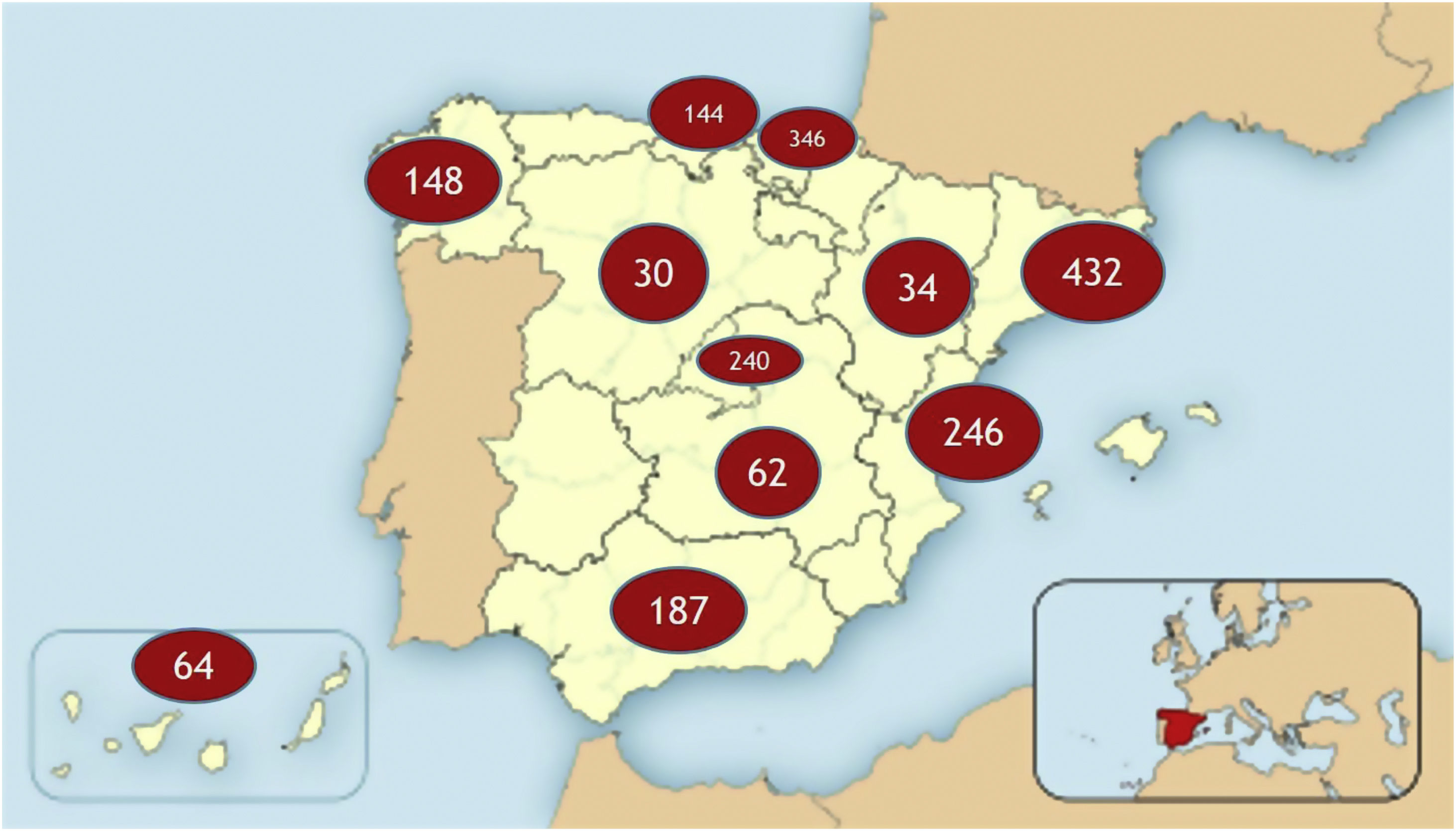
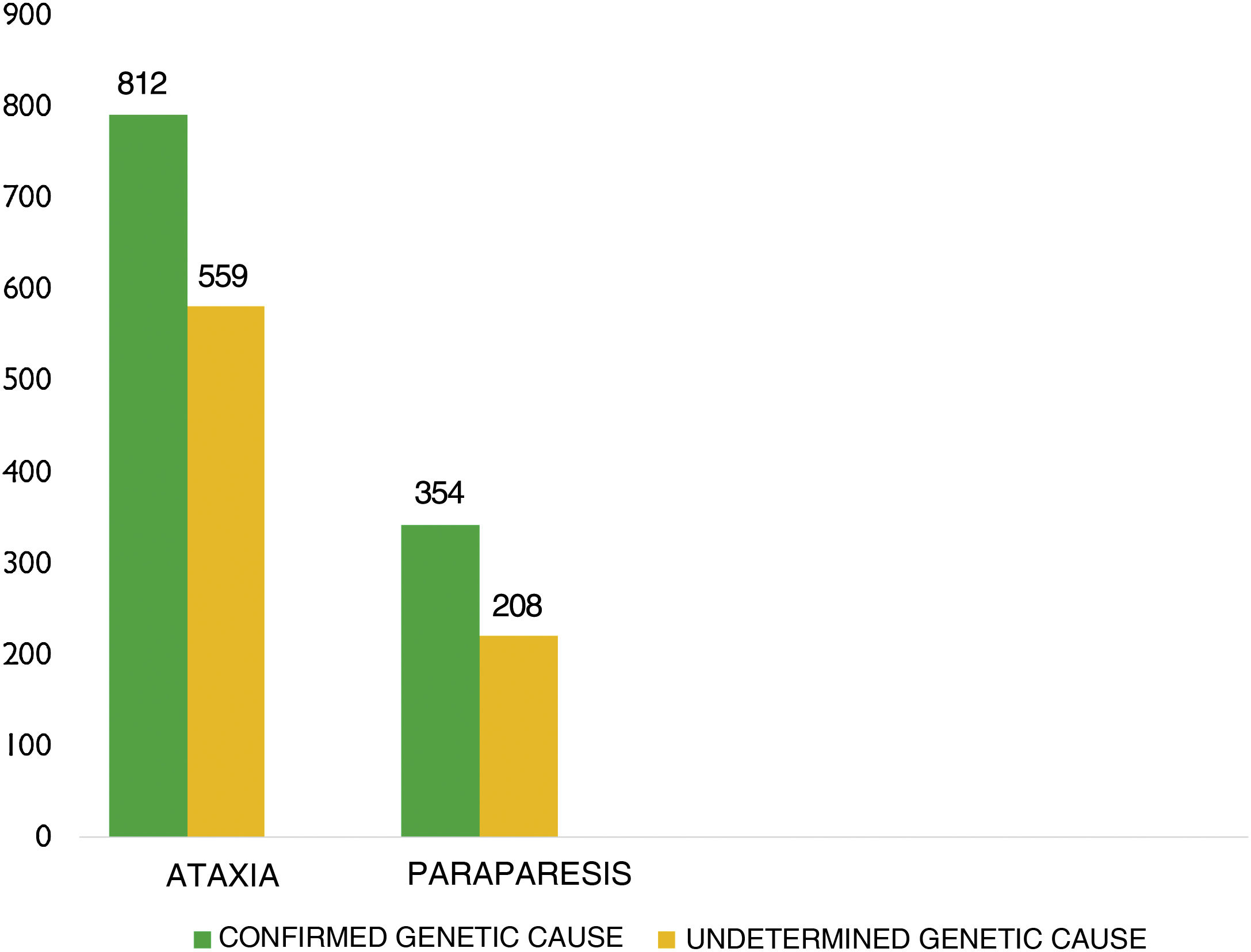
ResultsWe gathered data from a total of 1933 patients from 11 autonomous communities, provided by 47 neurologists or geneticists. Mean (SD) age in our sample was 53.64 (20.51) years; 938 patients were men (48.5%) and 995 were women (51.5%). The genetic defect was unidentified in 920 patients (47.6%). A total of 1371 patients (70.9%) had ataxia and 562 (29.1%) had hereditary spastic paraplegia. Prevalence rates for ataxia and hereditary spastic paraplegia were estimated at 5.48 and 2.24 cases per 100 000 population, respectively. The most frequent type of dominant ataxia in our sample was SCA3, and the most frequent recessive ataxia was Friedreich ataxia. The most frequent type of dominant hereditary spastic paraplegia in our sample was SPG4, and the most frequent recessive type was SPG7.
ConclusionsIn our sample, the estimated prevalence of ataxia and hereditary spastic paraplegia was 7.73 cases per 100 000 population. This rate is similar to those reported for other countries. Genetic diagnosis was not available in 47.6% of cases. Despite these limitations, our study provides useful data for estimating the necessary healthcare resources for these patients, raising awareness of these diseases, determining the most frequent causal mutations for local screening programmes, and promoting the development of clinical trials.
Las ataxias (AT) y paraparesias espásticas hereditarias (PEH) son síndromes neurodegenerativos raros. Nos proponemos conocer la prevalencia de las AT y PEH (APEH) en España en 2019.
Pacientes y métodosEstudio transversal, multicéntrico, descriptivo y retrospectivo de los pacientes con AT y PEH, desde Marzo de 2018 a Diciembre de 2019 en toda España.
ResultadosSe obtuvo información de 1.933 pacientes procedentes de 11 Comunidades Autónomas, de 47 neurólogos o genetistas. Edad media: 53,64 años ± 20,51 desviación estándar (DE); 938 varones (48,5%), 995 mujeres (51,1%). En 920 pacientes (47,6%) no se conoce el defecto genético. Por patologías, 1.371 pacientes (70,9%) diagnosticados de AT, 562 diagnosticados de PEH (29,1%). La prevalencia estimada de AT es 5,48/100.000 habitantes, y la de PEH es 2,24 casos/100.000 habitantes. La AT dominante más frecuente es la SCA3. La AT recesiva más frecuente es la ataxia de Friedreich (FRDA). La PEH dominante más frecuente es la SPG4, y la PEH recesiva más frecuente es la SPG7.
ConclusionesLa prevalencia estimada de APEH en nuestra serie es de 7,73 casos/100.000 habitantes. Estas frecuencias son similares a las del resto del mundo. En el 47,6% no se ha conseguido un diagnóstico genético. A pesar de las limitaciones, este estudio puede contribuir a estimar los recursos, visibilizar estas enfermedades, detectar las mutaciones más frecuentes para hacer los screenings por comunidades, y favorecer los ensayos clínicos.
Ataxia and hereditary spastic paraplegia (HSP) are neurodegenerative syndromes with a highly heterogeneous genetic basis. Their prevalence is unknown, but estimated at 3 to 20 cases per 100 000 population for ataxia and 1.2 to 9.6 cases per 100 000 population for HSP. Therefore, they are considered rare diseases.1–3
In Spain, the actual prevalence of both diseases remains unknown. Patient series from different autonomous communities and regions have been published over the past 2 decades; these studies include small, isolated samples.4–8 Some international multicentre studies have attempted to estimate the prevalence of these diseases, but with limitations.9–14 These studies report different prevalence rates due to differences in methodology, patient classification, and inclusion criteria. There are currently no screening programmes for population samples; prevalence studies are usually performed in areas with case clusters; and the published studies were mainly performed in European countries, with no prevalence studies from countries in North America, Africa, or Oceania.
An adequate epidemiological study requires exhaustive, “door-to-door” collection of data from each patient to obtain the largest number of cases and an optimal sample size, obtaining express consent from each patient for data collection and analysis of samples. It also entails the analysis of all the associated clinical and genetic variables, time progression, and the family relationship between the index patient and their descendants.
In our study, this methodology could not be used due to its complexity; therefore, we have accepted the limitations of a retrospective, cross-sectional design. This study estimates the prevalence of these diseases in Spain.
There is currently no epidemiological map of the types of ataxia and HSP in Spain. This information is essential for the development of healthcare policies, location and allocation of resources to reference units according to needs-based criteria, favouring care in specialised centres, identification of research strategies, and planning of clinical trials. More efficient management of resources would benefit the whole system, without losing focus on improving patient care.15 The aim of our study is to determine approximate prevalence data for ataxia and HSP in Spain in 2019, and the geographical distribution of the mutations and genes involved.
Material and methodsThis is a cross-sectional, multicentre, retrospective, descriptive, epidemiological study of the prevalence of ataxia and HSP in Spain in the year 2019 (diagnostic map). Our aim is to determine the prevalence of the different forms of ataxia and HSP and their distribution in the different Spanish autonomous communities. Data from patients without a genetic diagnosis are analysed separately.
We contacted the reference centres, units, and services for ataxia and hereditary paraplegia in Spain, and most ataxia units of the main hospitals in each autonomous community. Furthermore, through the Spanish Society of Neurology (SEN), we sent e-mails and placed notices on the SEN website to encourage the participation of neurologists, paediatric neurologists, and geneticists belonging to the society and attending patients with ataxia and HSP in Spain.
We included patients older than 16 years and younger than 60, recruited between March 2018 and December 2019, inclusive. Acquired cases were excluded.
The variables recorded were: age, sex, place and date of birth, date of symptom onset, attending centre, phenotype, family history, genetic study, and mutation (where possible). We also assessed cases with strong suspicion of hereditary origin, with or without family history, in whom genetic study results were negative, inconclusive, or indicated variants of uncertain significance.
The limitations of the study are those inherent to any cross-sectional, retrospective, descriptive, epidemiological study, in addition to those typical of a study of rare diseases. As data were anonymised, the risk of duplicates or overlapping data is one of the main selection biases that we may find. In an attempt to correct for this, we included the date of birth of every patient to control for duplicates. The limitations of the information sources are the available records and databases on these diseases at the hospitals and in the autonomous communities. In many cases, such resources do not exist, or are very rudimentary. The laboratory diagnostic techniques currently used are not comparable to those available 10 or 20 years ago, which may influence the number of cases with undetermined genetic origin.
Data were anonymised and grouped, and were stored in a Microsoft Excel spreadsheet and subsequently processed with the SPSS software, version 25 (IBM Statistics), for the statistical analysis. For the estimated prevalence rate, we considered that, as of 1 January 2019, Spain had a population of 47 007 367, according to census data from the Spanish National Statistics Institute.16 Of this total, 23 033 803 were men and 23 973 564 were women. In our calculations, we included figures for the population between the ages of 16 and 60 years (25 017 427 million people).
This project was approved by the SEN’s Ethics and Deontology Committee (February 2018) and by the different Ethics Committees of most autonomous communities.
ResultsWe included a total of 1933 patients from 11 autonomous communities, with the participation of hospitals of different sizes, national reference centres, units, and services for ataxia and hereditary paraplegia, and a total of 47 neurologists or geneticists (Fig. 1). The total estimated prevalence of ataxia and HSP amounts to 7.73 cases/100 000 population.

Mean age (SD) was 53.64 (20.51) years (median, 54). Of the total sample, 938 patients were men (48.5%) and 995 were women (51.5%). The underlying genetic defect was unknown in 920 patients (47.6%) (Fig. 2).

By disease, 1371 patients (70.9%) had been diagnosed with ataxia and 562 (29.1%) with HSP (Table 1).
Subtypes of ataxia and HSP in our series (index cases in parentheses).
| N | Dominant ataxia | Recessive ataxia | Other ataxias | Dominant HSP | Recessive HSP | Other HSP |
|---|---|---|---|---|---|---|
| SCA, 131 cases (47) | FRDA, 249 cases (232) | FXTAS, 20 cases (19) | SPG4, 150 cases (80) | SPG7, 56 cases (41) | ABCD1, 11 cases (11) | |
| SCA2, 47 cases (36) | NPC1, 18 cases (16) | Pelizaeus-Merzbacher disease, 1 case (1) | SPG17, 32 cases (10) | SPG11, 22 cases (21) | 5PG2, 1 case (1) | |
| SCA6, 37 cases (27) | AOA2, 21 cases (13) | Kearns-Sayre syndrome, 1 case (1) | SPG10, 21 cases (13) | SPG5A, 5 cases (5) | ||
| SCA1, 37 cases (29) | AT-T, 17 cases (12) | SPG3A, 14 cases (12) | SPG15, 4 cases (4) | |||
| SCA37, 34 cases (14) | SPAX6, 12 cases (9) | SPG31, 11 cases (8) | SPG35, 4 cases (4) | |||
| AE2, 35 cases (25) | Cerebrotendinous xanthomatosis, 12 cases (8) | SPG13, 3 cases (2) | SPG48, 4 cases (2) | |||
| SCA36, 24 cases (15) | SCAR8/ARCA1, 9 cases (7) | ODD, 2 cases (2) | SPG76, 4 cases (3) | |||
| SCA7, 15 cases (14) | POLR3A, 6 cases (5) | SPG12, 1 case (1) | SPG54, 3 cases (1) | |||
| SCA48, 7 cases (1) | SCAR10, 6 cases (3) | SPG20, 2 cases (1) | ||||
| SCA17, 6 cases (6) | Vitamin E deficiency, 5 cases (5) | SPG30, 2 cases (1) | ||||
| SCA19, 6 cases (3) | POLG1, 4 cases (4) | SPG43, 1 case (1) | ||||
| AE1, 6 cases (6) | AOA4, 3 cases (3) | SPG46, 1 case (1) | ||||
| SCA8, 5 cases (5) | SPAX4, 2 cases (1) | |||||
| SCA23, 2 cases (2) | Vanishing white matter disease, 2 cases (1) | |||||
| SCA35, 2 cases (2) | Cockayne syndrome A, 2 cases (1) | |||||
| SCA40, 2 cases (1) | Cockayne syndrome B, 2 cases (1) | |||||
| DRPLA, 2 cases (2) | SPAX2, 1 case (1) | |||||
| Alexander disease, 2 cases (2) | SPAX5, 1 case (1) | |||||
| SCA13, 1 case (1) | PCARP, 1 case (1) | |||||
| SCA12, 1 case (1) | SCAR16, 1 case (1) | |||||
| SCA10, 1 case (1) | Homocysteine methylation deficit, 1 case (1) | |||||
| OPA1, 1 case (1) | Wilson disease, 1 case (1) | |||||
| AE5, 1 case (1) | Sandhoff, 1 case (1) | |||||
| Cerebellar ataxia+hemiplegic migraine, 1 case (1) | POLR3B, 1 case (1) | |||||
| PMM2, 1 case (1) | ||||||
| Brown–Vialetto–Van Laere syndrome, 1 case (1) | ||||||
| Marinesco–Sjögren syndrome, 1 case (1) | ||||||
| TOTAL | 406 (294) | 384 (336) | 22 (21) | 234 (171) | 108 (85) | 12 (12) |
AT-T: ataxia telangectasia.
The estimated prevalence of ataxia in our series, according to the data from the participating communities, amounts to 5.48 cases per 100 000 population (dominant ataxia in 1.62 cases/100 000 population and recessive ataxia in 1.53 cases/100 000 population).
The estimated prevalence of HSP in the same age group amounts to 2.24 cases per 100 000 population (estimated prevalence rates of 0.93 cases/100 000 population for dominant HSP and 0.43 cases/100 000 population for recessive HSP) (Table 2).
Estimated prevalence in the published series.
| Ataxia and HSP | HSP | Ataxia | Other | |
|---|---|---|---|---|
| Ruano13 (global) | 9.8/105 | 3.6/105 | 6/105 | – |
| **Polo4 (Cantabria) | 20.2/105 (index: 9.4/105) | 9.6/105 | 10.6/105 | – |
| **Infante5 (Cantabria) | – | – | – | AD-HA: 1.6/105 |
| Erichsen, 2009 (Norway) | 13.9/105 | 7.4/105 | 6.5/105 | – |
| Braschinsky, 2009 (Estonia) | – | 4.4/105 | – | – |
| Orsucci, 2014 (Tuscany) | – | 2.2−3.4/105 | – | – |
| Hutchinson, 2002 (Ireland) | – | – | – | “Pure” HSP-DA: 1.27/105 |
| Zortea, 2004 (Padua) | – | – | 9.3/105 | – |
| Coutinho, 2013 (Portugal) | 13/105 | 4.1/105 | 8.9/105 | – |
| Present study | 7.73/105 | 5.48/105 | 2.24/105 |
By subtype, and exclusively assessing cases with confirmed genetic cause: of the 406 cases with dominant ataxia, the most frequent types were SCA3 (131 cases) and SCA2 (47 cases). Of the 384 patients with recessive ataxia, the most frequent forms were Friedrich ataxia (249 cases) and NPC1 (18 cases). Regarding HSP, of the 234 patients with dominant forms, the most frequent were SPG4 (150 patients) and SPG17 (32); of the 108 cases of recessive HSP, the most frequent were SPG7 (56) and SPG11 (22) (Table 1).
DiscussionDetermining the prevalence of rare diseases is a challenge for any healthcare system or population group.17 Their low prevalence, geographical dispersion, and high diagnostic complexity, together with the difficulty of accessing the necessary tests, especially genetic tests, result in limited visibility of these diseases and difficulty quantifying them.18–27
This study sought to overcome some of these difficulties to determine the prevalence of ataxia and HSP in Spain.
The initiative was launched in 2017 by the SEN’s Commission for the Study of Ataxias and Degenerative Spastic Paraplegias, which planned the creation of a cross-sectional map to estimate the prevalence of ataxia and HSP in Spain. Over the following 2 years, the SEN and the Commission for the Study of Ataxias and Degenerative Spastic Paraplegias have made an organisational effort to make contact with and to diffuse this project to the largest number of neurologists possible, as well as geneticists and paediatric neurologists. This screening study presents clear limitations associated with the study design, the difficulty of organising a multicentre study, and the exclusion of patients younger than 16 years. However, we believe that the generous collaboration of the participating neurologists, the access to them through the e-mail sent by the SEN, the simple design of the registry, and the coordinated joint effort have led to the collection of a large body of valuable data that may be compared against previous findings in the Spanish setting and series from other countries.
Thus, in Spain, the first attempt to perform a multicentre study of these diseases was the Spanish Registry of Ataxia and Degenerative Spastic Paraparesis, launched in 2012 by a group of Spanish experts with the support of the Research Institute for Rare Diseases of the Institute of Health Carlos III, with rigorous and ambitious goals. This project is currently suspended due to several factors, and we are unfortunately unable to analyse its results or access them to compare them with our own.
Five Spanish series have been published to date,4–8 3 of which were performed in Cantabria; in 1991, Polo et al.4 published a series with 48 index cases of ataxia and HSP registered only in this autonomous community in approximately 10 years. That study mainly reported 18 families with Friedreich ataxia and 9 with HSP. They described a prevalence of ataxia and HSP of 20.2 cases per 100 000 population. The 1999 study by Pujana et al.7 described a series of 87 families, and 60 sporadic cases, only reporting cases of ataxia (from 9 autonomous communities); SCA2 and SCA3 were present in 15% of patients, and 58% were genetically unclassifiable. The series by Infante et al.5 focused on autosomal dominant ataxias in Cantabria, reporting 30 families with mainly SCA2 and SCA3; they did not find a specific mutation in 9 families (30%). The estimated prevalence of this group of ataxias amounts to 1.6 cases per 100 000 population. The Spanish series by Mayo Cabrero et al.,8 from the year 2000, describes 121 patients with dominant ataxia and Friedreich ataxia, aged between 5 and 80 years, from 8 autonomous communities. The most frequent dominant ataxia was SCA3, followed by SCA7, whereas SCA2 was much less frequent. No genetic cause was identified in 41.2% of cases of dominant ataxias and 43.5% of cases of recessive ataxias.
When comparing our findings with those from these series, we found 2 significant limitations: only one article included both ataxia and HSP; and, as they were published more than 20 years ago, genetic testing and diagnostic and recording methods have significantly changed, hindering comparison.
Regarding global data, a systematic review published in 2014 included 22 studies published between 1983 and 2013, with a total of 14 539 patients from 16 different countries.13 This extensive review reported estimated prevalence rates of 5.6 cases per 100 000 population for dominant ataxia, and 3.3 cases per 100 000 population for recessive ataxia (not all cases were genetically confirmed). The estimated prevalence in our series is 1.62 cases per 100 000 population for dominant ataxia, and 1.53 cases per 100 000 population for recessive ataxia. Regarding the prevalence of HSP, the systematic review described a prevalence of autosomal dominant HSP of 5.5 cases per 100 000 population, and autosomal recessive HSP of 5.3 cases per 100 000 population, vs 0.93 cases and 0.43 cases per 100 000 population, respectively, in our series.
Despite the significant number of cases gathered, the lower prevalence in our series compared with other published series, both for ataxias and HSP, may be due to several factors. One of these is the limited age range we established to collect data (from 16 to 59 years: we excluded paediatric cases, and those older than 60 years, due to the possible copresence of such other causes of ataxia as cerebrovascular disease, tumour, etc). Although we achieved excellent collaboration and communication with most autonomous communities, some were unable to participate, which has limited the total number of cases gathered. Local factors, such as the limited access to genetic testing, or differences in patient access to the resources and units or specialists in their respective autonomous communities, may also have been limiting factors.
If we analyse the prevalences of the different subgroups in our series, SCA3 was the most frequent autosomal dominant ataxia, whereas Friedreich ataxia was the most prevalent recessive form, similarly to the data reported in previous publications. However, the high numbers of patients with SCA36, SCA37, and NPC1 identified in our study are surprising. These differences are explained by the fact that SCA36 and SCA37 were originally described in Spain, which has led to the identification of a larger number of cases during the initial research and to a subsequent awareness of this diagnosis in the Spanish geographical areas where they were described. Regarding NPC1, we believe that the figures are related to the specific interest of some local groups in this disease, which has led to the identification of a greater number of cases in those centres that have actively collaborated in the registry.
Regarding paraparesis, the 2014 review13 reported that the most frequent dominant form was SPG4, followed by SPG3; and among recessive HSPs, the most frequent was SPG11, followed by SPG15. These data are similar to those reported in other international publications with smaller samples.8,9 In our series, SPG4 was the most frequent dominant form, and among recessive forms, SPG7 was the most prevalent; this is consistent with those publications. In turn, SPG15 was the fourth most frequent form in our series, with only 4 identified index cases.
Finally, the study has shown that 47.6% of our patients with ataxia and HSP lack a definitive genetic diagnosis. This figure is within the range published for these entities (33%–92%),18–28 and may be improved with better implementation of genetic testing.
ConclusionsOur study estimates the prevalence of ataxia and HSP in Spain at 5.48 and 2.24 cases per 100 000 population, respectively. The most frequent type of dominant ataxia is SCA3, followed by SCA2. The most frequent recessive ataxias are Friedreich ataxia and Niemann-Pick disease type C. The most frequent type of dominant HSP is SPG4, followed by SPG17, and the most frequent recessive type is SPG7, followed by SPG11.
These rates are similar to those reported for other countries. Genetic diagnosis was unavailable in 47.6% of cases. Despite these limitations, this is the best information available on the prevalence, frequency, and geographical distribution of these diseases in Spain.
Genetic diagnosis is essential to improve everyday medical care, to raise awareness of these diseases, to detect the most frequent mutations with a view to designing more appropriate screening programmes for an area or community, to optimise national registries, to assess the need for resources, to design clinical trials, and to facilitate the recruitment of patients to be included in the study.
FundingThe results of our study were partially presented at the 2017 and 2018 Annual Meetings of the Spanish Society of Neurology. Our study was partially funded by a grant from the Spanish Society of Neurology awarded to the lead author (Dr Gloria Ortega Suero), who was responsible for the database and data custody.
Conflicts of interestThe authors have no conflicts of interest to declare.
