



Autonomic dysfunction is a common manifestation of hereditary transthyretin (ATTRv) amyloidosis that frequently appears early and has a major impact on disease severity, survival, and quality of life. Early detection of autonomic symptoms is paramount to avoid delayed diagnosis or misdiagnosis of ATTRv amyloidosis and thereby achieve timely interventions with novel therapies. We thus require a better understanding and recognition of the warning signs of autonomic nervous system involvement in patients with ATTRv amyloidosis.
DevelopmentAccording to the literature and the expertise of key opinion leaders on ATTRv amyloidosis, early signs and symptoms of autonomic dysfunction often include orthostatic hypotension, gastrointestinal disturbances, and cardiac and sudomotor sympathetic denervation. Assessment methods for a rapid and accurate diagnostic thus comprise blood pressure monitoring, nutritional status evaluation, cardiac imaging techniques, and pathological and neurophysiological tests assessing small fiber function. The COMPASS-31 and Norfolk QOL-DN tests allow assessment of the severity of autonomic dysfunction and its impact on quality of life, respectively.
ConclusionThe screening strategy for autonomic dysfunction should involve diverse neurological and non-neurological tests to encompass the wide variety of potential manifestations of autonomic dysfunction in ATTRv amyloidosis. Further studies addressing the correlations among different tests and their prognostic value in different TTR variants are required to establish a single standardized method or the minimum set of tests needed to assess autonomic dysfunction in ATTRv amyloidosis patients.
La disfunción autonómica es una manifestación común de la amiloidosis hereditaria por transtiretina (ATTRv) que con frecuencia aparece temprano y tiene un impacto importante en la gravedad de la enfermedad, la supervivencia y la calidad de vida. La detección temprana de los síntomas autonómicos es fundamental para evitar el diagnóstico tardío o el diagnóstico erróneo de la ATTRv amyloidosis y, por lo tanto, lograr intervenciones oportunas con terapias novedosas. Por lo tanto, requerimos una mejor comprensión y reconocimiento de los signos de advertencia de la participación del sistema nervioso autónomo en pacientes con ATTRv amiloidosis.
DesarrolloDe acuerdo con la literatura y la experiencia de líderes de opinión clave sobre la ATTRv amiloidosis, los primeros signos y síntomas de disfunción autonómica a menudo incluyen hipotensión ortostática, trastornos gastrointestinales y denervación simpática cardíaca y sudomotora. Los métodos de evaluación para un diagnóstico rápido y preciso comprenden, por lo tanto, el control de la presión arterial, la evaluación del estado nutricional, las técnicas de imagen cardíaca y las pruebas patológicas y neurofisiológicas que evalúan la función de las fibras pequeñas. Las pruebas COMPASS-31 y Norfolk QOL-DN permiten evaluar la gravedad de la disfunción autonómica y su impacto en la calidad de vida, respectivamente.
ConclusiónLa estrategia de detección de disfunción autonómica debe incluir diversas pruebas neurológicas y no neurológicas para abarcar la amplia variedad de posibles manifestaciones de disfunción autonómica en la ATTRv amiloidosis. Podemos proporcionar recomendaciones tempranas para el manejo de la disautonomía en este contexto, basándose no solo en nuestra revisión de la literatura, sino también en nuestra experiencia clínica como expertos líderes en el campo. Sin embargo, se requieren más estudios que aborden las correlaciones entre diferentes pruebas y su valor pronóstico en diferentes variantes de TTR para establecer un único método estandarizado o el conjunto mínimo de pruebas necesarias para evaluar la disfunción autonómica en pacientes con ATTRv amiloidosis.
Hereditary transthyretin (ATTRv) amyloidosis is a rapidly progressive disease caused by mutations in the transthyretin (TTR) gene. The resulting misfolded TTR proteins are deposited as amyloid fibrils in different body tissues, including the nerves, heart, gastrointestinal tract, kidneys, eyes, and connective tissue.1,2 Although ATTRv amyloidosis disease phenotypes have traditionally been classified as neuropathic or cardiac according to the predominant clinical manifestation, most patients develop a mixed phenotype with varying degrees of neuropathic and cardiac involvement, as well as other manifestations in other organs, which is why it is considered a multisystem disease.3
Autonomic dysfunction is present in 50–80% of patients with ATTRv amyloidosis and has a major impact on disease severity, survival, and quality of life (QoL).4 However, symptoms related to autonomic dysfunction are nonspecific and not always spontaneously reported by patients. Hence, their prevalence in ATTRv amyloidosis may be underestimated. As the disease progresses, the diagnosis of autonomic disturbances may be hampered by severe peripheral neuropathy and/or cardiomyopathy.5
Although autonomic dysfunction can affect patients with any TTR variant, early-onset V30M patients are more prone to autonomic manifestations,6 which are often one of the first signs in these patients.7 In contrast, autonomic symptoms are mild in patients with the V30M variant and late-onset ATTRv amyloidosis and generally appear later in the course of the disease.4,8–10 Autonomic symptoms are also common in non-V30M patients and can include both cardiac and peripheral vasomotor autonomic dysfunction.11 The prevalence of gastrointestinal manifestations, which are mostly related to autonomic dysfunction, was as high as 56% in non-V30M patients in the THAOS registry (the first global Transthyretin Amyloidosis Outcomes Survey). However, the prevalence of gastrointestinal disturbances was lower and similar to that seen in the general population in patients carrying TTR variants associated with a predominantly cardiac phenotype.12
Autonomic dysfunction typically appears at onset or in the early stages of ATTRv amyloidosis, at least in patients with early-onset disease. Because patients with ATTRv amyloidosis are more prone to autonomic symptoms than patients with other types of amyloidosis or other adult-onset progressive neuropathies,4 these symptoms are considered some of the “red flag” symptoms for diagnosis and should raise suspicion of ATTRv amyloidosis.1 Recognition of the symptoms of the disease and its early diagnosis are now more important than ever due to the advent of novel therapies for this disease. These new treatment options, such as TTR stabilizers and TTR gene silencers with the ability to slow or delay disease progression, are particularly effective in early disease stages.13–15 Due to the high variability of autonomic manifestations among patients with pathogenic TTR variants, a better understanding and recognition of the warning signs of autonomic nervous system involvement is crucial to avoid a delayed diagnosis or misdiagnosis that may lead to inappropriate interventions. This review provides a comprehensive overview of the most relevant autonomic symptoms in patients with ATTRv amyloidosis, along with the most common methods for assessing autonomic function. The advantages and limitations of the available tests are discussed to improve the evaluation of autonomic dysfunction in patients with TTR variants.
MethodologyKey opinion leaders in ATTRv amyloidosis met to discuss the importance of autonomic dysfunction in this disease and the different approaches currently used to evaluate the heterogeneous manifestations of autonomic dysfunction in patients with ATTRv amyloidosis. Two remote meetings were conducted: an advisory board in September 2020 and a review meeting in October 2020.
To provide an overview of autonomic symptoms in patients with pathogenic TTR variants and the most useful tests for their early diagnosis, a comprehensive literature search was performed using the PubMed database. Related publications were identified by searching for the following terms: hereditary transthyretin amyloidosis, ATTRv, hATTR, transthyretin familial amyloid polyneuropathy, transthyretin amyloid neuropathy, autonomic function, dysautonomia, autonomic neuropathy, or autonomic dysfunction, and assessment, diagnosis, testing, analysis, measures, management, or treatment.
Based on a literature review, as well as the clinical experience of the key opinion leaders and the assessments that they routinely use in clinical practice, this article aims to provide clinicians with a better understanding of autonomic manifestations and their significance in the diagnosis and prognosis of ATTRv amyloidosis. The reported data on the advantages and limitations of the different assessments and the correlations among them are discussed to facilitate the selection of a battery of tests for the rapid and accurate detection of autonomic dysfunction, which should support the early detection of ATTRv amyloidosis.
ResultsEtiopathogenesis and pathophysiology of autonomic dysfunctionAutonomic dysfunction can be a clinical manifestation of several underlying diseases, such as diabetes, Parkinson disease, chronic kidney disease, and amyloidosis.5,16 Although autonomic disturbances caused by the accumulation of abnormal amyloid deposits in the autonomic nervous system can occur in all types of amyloidosis, including amyloid light-chain amyloidosis and serum amyloid A protein amyloidosis, they are more frequent in ATTRv amyloidosis.5 The amyloid deposition of TTR initially affects small nerve fibers, which are the main constituents of both the sympathetic and parasympathetic nerves of the autonomic nervous system. In turn, sympathetic nerve impairment may affect the noradrenergic, adrenergic, or cholinergic subsystems, which differ in function and in main chemical messenger. The sympathetic noradrenergic system is involved in cardiovascular regulation, the adrenergic system plays a major role in the maintenance of metabolic homeostasis in response to stress, and the cholinergic system is crucial for the regulation of sweat secretion.17 Due to its extensive nature, any alterations to the autonomic nervous system are associated with a wide range of clinical manifestations that can affect nearly any system. Thus, patients with autonomic dysfunction may present a variety of apparently unrelated and hard-to-diagnose symptoms. Manifestations of autonomic dysfunction with a major impact on disability and QoL include orthostatic hypotension, gastrointestinal motility disorders, sexual dysfunction, anhidrosis, and neurogenic bladder.7
Cardiovascular autonomic dysfunction due to impaired sympathetic and parasympathetic control of the cardiovascular system is the most threatening feature of autonomic dysfunction because it can lead to arrhythmias and sudden death.18 The most common manifestation of cardiovascular autonomic dysfunction in patients with ATTRv amyloidosis is orthostatic hypotension.7,16,19 When symptomatic, orthostatic hypotension can present as dizziness, lightheadedness, blurred vision when standing, falls, and syncope.20,21
Gastrointestinal dysfunction is common in ATTRv amyloidosis, particularly in patients with the V30M variant. According to the enrollment data collected in the THAOS registry, the most common gastrointestinal disturbances in ATTRv amyloidosis patients are unintentional weight loss, early satiety, and alternating diarrhea/constipation, followed by constipation, diarrhea, nausea, vomiting, and fecal incontinence.12
Bladder dysfunction caused by damage to the parasympathetic fibers starts with urinary retention, which increases the risk of urinary tract infections, and may progress to urinary incontinence.5 One of the first signs of ATTRv amyloidosis in men is erectile dysfunction proceeding to impotence.
Other manifestations of autonomic dysfunction include miosis at rest and scalloped pupils, which are caused by pupillomotor dysfunction and, unlike the symptoms described above, are usually asymptomatic.20 Further symptoms may include accommodative dysfunction with blurred vision and sensitivity to light.
Accordingly, early diagnosis of autonomic dysfunction requires the correct association of these diverse and sometimes nonspecific symptoms with a single underlying disease.
Neurological tests to assess autonomic dysfunctionVarious tests can be used to assess autonomic function. Many are composite tests that also explore other symptoms.
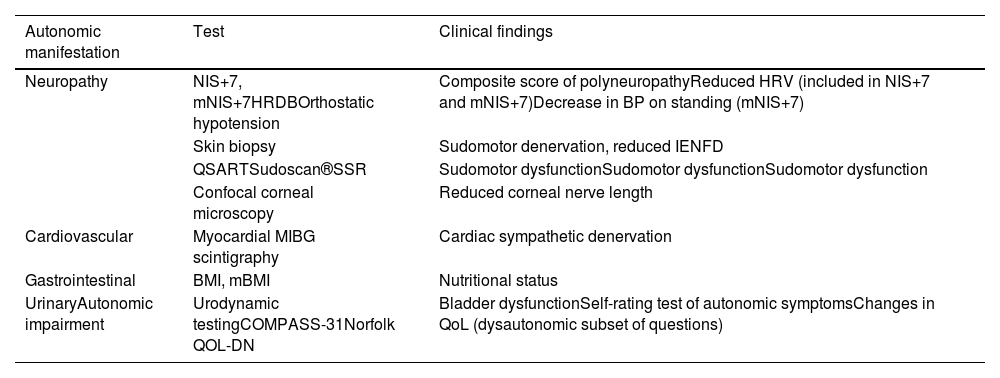
Neuropathy Impairment Score Plus 7 and modified Neuropathy Impairment Score Plus 7Autonomic dysfunction is one of the components of both the Neuropathy Impairment Score Plus 7 (NIS+7) and modified Neuropathy Impairment Score Plus 7 (mNIS+7) scales, commonly used in clinical trials. The NIS+7 is a composite score of polyneuropathy that combines the weakness, reflexes, and sensation measures of the Neuropathy Impairment Score (NIS) with nerve conduction studies, sensory measures, and autonomic dysfunction assessed by heart rate response to deep breathing (HRDB) (Table 1), allowing a better characterization and quantification of neuropathic impairment.22 In response to the limitations of the NIS+7 in evaluating small sensory fiber dysfunction in ATTRv amyloidosis patients, the mNIS+7 incorporates Smart Somatotopic Quantitative Sensory Testing (S ST QST), which can accurately estimate the body surface distribution of sensory loss.22
Assessments and tests for evaluating autonomic dysfunction.
| Autonomic manifestation | Test | Clinical findings |
|---|---|---|
| Neuropathy | NIS+7, mNIS+7HRDBOrthostatic hypotension | Composite score of polyneuropathyReduced HRV (included in NIS+7 and mNIS+7)Decrease in BP on standing (mNIS+7) |
| Skin biopsy | Sudomotor denervation, reduced IENFD | |
| QSARTSudoscan®SSR | Sudomotor dysfunctionSudomotor dysfunctionSudomotor dysfunction | |
| Confocal corneal microscopy | Reduced corneal nerve length | |
| Cardiovascular | Myocardial MIBG scintigraphy | Cardiac sympathetic denervation |
| Gastrointestinal | BMI, mBMI | Nutritional status |
| UrinaryAutonomic impairment | Urodynamic testingCOMPASS-31Norfolk QOL-DN | Bladder dysfunctionSelf-rating test of autonomic symptomsChanges in QoL (dysautonomic subset of questions) |
NIS+7: Neuropathy Impairment Score Plus 7; mNIS+7: modified Neuropathy Impairment Score Plus 7; HRDB: heat rate response to deep breathing; BP: blood pressure; IENFD: intraepidermal nerve fiber density; QSART: quantitative sudomotor axon reflex test; SSR: sympathetic skin response; BMI: body mass index; mBMI: modified body mass index; COMPASS-31: Composite Autonomic Symptom Score; QoL, quality of life.
The mNIS+7 can also differ from the NIS+7 in terms of autonomic testing. Whereas the NIS+7 uses HRDB as the only measure of autonomic dysfunction, the mNIS+7 can use HRDB or orthostatic hypotension (Table 1). The latter was introduced in response to the limitations of HRBD in patients with arrhythmias or pacemakers.23
HRDB assesses heart rate variability (HRV), which can be reduced in ATTRv amyloidosis patients. The HRBD test is performed by continuously recording the electrocardiogram and the breathing pattern during several cycles of controlled deep breathing. In the mNIS+7, the HRDB score ranges from normal (0 points) to very abnormal (3.7 points) based on normal deviates from percentile reference values.22,24 In contrast, the use of orthostatic hypotension as a measure of autonomic dysfunction is based on the decrease in blood pressure on standing compared with that on sitting or lying down. Decreases of 20mmHg or more in systolic blood pressure or 10mmHg or more in diastolic blood pressure indicate orthostatic hypotension.19,21 For its evaluation, patients are asked to sit or lie down for 5min before standing or being passively tilted upright for 1–3min while systolic blood pressure, diastolic blood pressure, and heart rate are recorded. An additional recording at 5min can be included but is more useful in other diseases such as Parkinson disease.25 By comparison with reference values, the orthostatic hypotension score in the mNIS+7 (0–2 points) depends on whether the function is classified as normal (<95th percentile), mildly reduced (95th to <99th percentile), or very reduced (≥99th percentile). A higher score indicates a worse degree of orthostatic hypotension.
Small fiber testingThe abnormal functioning of the autonomic sympathetic fibers innervating sweat glands is one of the earliest findings in small fiber neuropathy and therefore a good indicator of autonomic dysfunction. Sweat glands and epidermal innervation can be pathologically examined using skin biopsy. Thus, evidence of sudomotor denervation and a reduced intraepidermal nerve fiber density (IENFD) obtained by immunostaining of skin sections is indicative of small fiber neuropathy26 (Table 1). However, this technique is available in only some laboratories.
In addition, several neurophysiological tests can be applied to assess sudomotor dysfunction, including the quantitative sudomotor axon reflex test (QSART), Sudoscan®, and sympathetic skin response (SSR) (Table 1). The QSART evaluates sympathetic cholinergic sudomotor function by measuring the amount of sweat produced upon stimulation of the sweat glands by iontophoresis of a cholinergic agent. Abnormal sweat production indicates small fiber damage.27 Assessment of sudomotor function by the Sudoscan is based on the use of mild electrical stimulation and reverse iontophoresis to measure the electrochemical skin conductance, which depends on sweat chloride production and its reaction with the nickel in the electrodes. A low electrochemical skin conductance in the Sudoscan is indicative of sudomotor dysfunction.28,29 Whereas both the QSART and Sudoscan directly assess sympathetic cholinergic sudomotor function, the SSR is an indirect evaluation based on changes in the skin potential generated by sweat glands and the epidermis after rapid inspiration or electrical stimulation.27 A proposal for the use of these tools for idiopathic neuropathy (Table 2) and ATTRv amyloidosis (Table 3) is presented in this article.
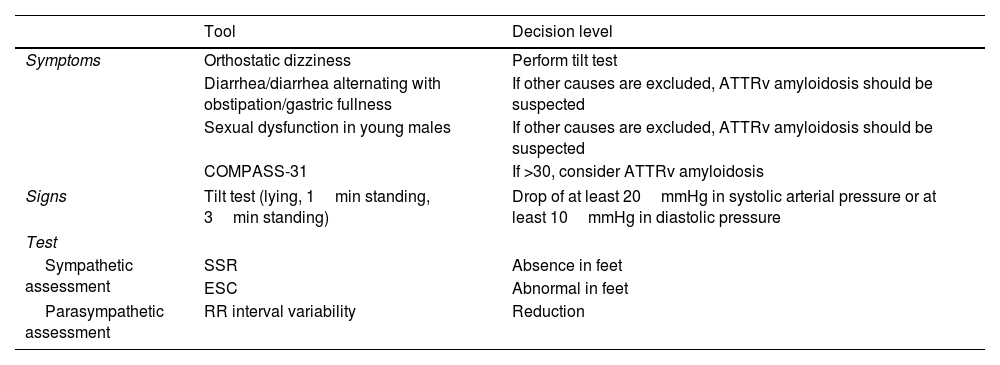
Dysautonomic assessment for the diagnosis of ATTRv amyloidosis in patients presenting with “idiopathic neuropathy”.
| Tool | Decision level | |
|---|---|---|
| Symptoms | Orthostatic dizziness | Perform tilt test |
| Diarrhea/diarrhea alternating with obstipation/gastric fullness | If other causes are excluded, ATTRv amyloidosis should be suspected | |
| Sexual dysfunction in young males | If other causes are excluded, ATTRv amyloidosis should be suspected | |
| COMPASS-31 | If >30, consider ATTRv amyloidosis | |
| Signs | Tilt test (lying, 1min standing, 3min standing) | Drop of at least 20mmHg in systolic arterial pressure or at least 10mmHg in diastolic pressure |
| Test | ||
| Sympathetic assessment | SSR | Absence in feet |
| ESC | Abnormal in feet | |
| Parasympathetic assessment | RR interval variability | Reduction |
SSR: sympathetic skin response; ESC: electrochemical skin conductance.
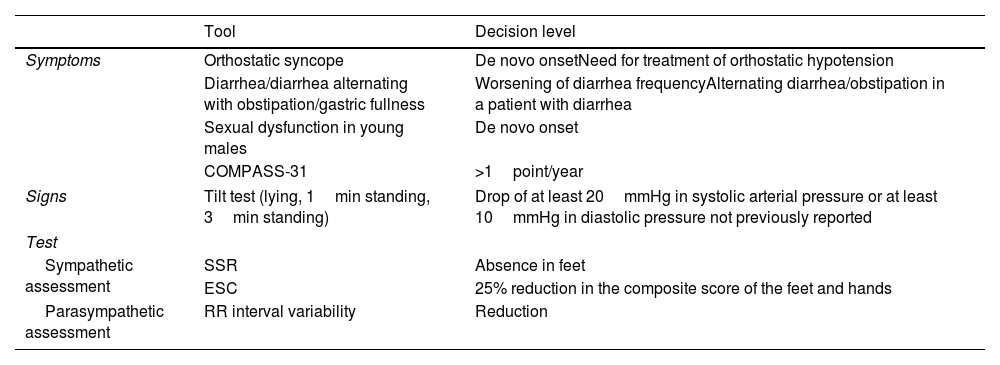
Dysautonomia for the follow-up of ATTRv amyloidosis patients.
| Tool | Decision level | |
|---|---|---|
| Symptoms | Orthostatic syncope | De novo onsetNeed for treatment of orthostatic hypotension |
| Diarrhea/diarrhea alternating with obstipation/gastric fullness | Worsening of diarrhea frequencyAlternating diarrhea/obstipation in a patient with diarrhea | |
| Sexual dysfunction in young males | De novo onset | |
| COMPASS-31 | >1point/year | |
| Signs | Tilt test (lying, 1min standing, 3min standing) | Drop of at least 20mmHg in systolic arterial pressure or at least 10mmHg in diastolic pressure not previously reported |
| Test | ||
| Sympathetic assessment | SSR | Absence in feet |
| ESC | 25% reduction in the composite score of the feet and hands | |
| Parasympathetic assessment | RR interval variability | Reduction |
SSR: sympathetic skin response; ESC: electrochemical skin conductance.
Finally, confocal corneal microscopy is a rapid and noninvasive technique allowing the visualization and quantification of corneal nerves in vivo (Table 1). Corneal nerve fiber length measured by in vivo confocal microscopy is reduced in patients with ATTRv amyloidosis.30 Again, this last technique is not widely available.
Non-neurological testsPatient-reported outcomesThe Composite Autonomic Symptom Score (COMPASS-31) is a self-rating questionnaire for assessing symptoms of autonomic dysfunction (Table 1). It contains 31 items evaluating the following six autonomic domains: orthostatic hypotension (40 points), vasomotor (5 points), secretomotor (15 points), gastrointestinal (25 points), bladder (10 points), and pupillomotor (5 points). Thus, the COMPASS-31 is graded on a 100-point scale, with higher scores indicating greater autonomic impairment. Its main pitfall is that it does not consider sexual dysfunction. Because the COMPASS-31 is a complex and time-consuming questionnaire, shorter and easier scoring systems have been developed. For instance, the Compound Autonomic Dysfunction Test (CADT) is a 4-item questionnaire that evaluates the main autonomic symptoms of ATTRv amyloidosis, including orthostatic hypotension, nausea and vomiting, diarrhea/constipation, and sphincter disturbances.31 In males, an additional item evaluates sexual dysfunction. Each item is graded from 0 to 4, with lower scores indicating greater impairment. The Survey of Autonomic Symptoms (SAS) can also be useful for assessing autonomic dysfunction. It contains 11 items evaluating the following autonomic domains: orthostatic hypotension, sudomotor, vasomotor, gastrointestinal, and urinary.32 As in the CADT, an additional question evaluates sexual dysfunction in male patients. The SAS score is based on the presence of symptoms and their severity, which is graded from 1 (mild) to 5 (severe), with higher scores therefore indicating greater impairment.
The Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QOL-DN) is a 35-item self-rating QoL questionnaire (Table 1). Although the Norfolk QOL-DN was initially developed to evaluate QoL in patients with diabetic neuropathy, it has also proved useful for assessing the impact of ATTRv amyloidosis on QoL.33 In particular, one of the five evaluated domains refers to autonomic dysfunction. Scoring is based on the composite score of the five domains: large fiber neuropathy (56 points), symptoms (32), activities of daily living (20), small fiber neuropathy (16), and autonomic neuropathy (12). The maximum impairment score is thus 136 points, with a higher score indicating worse QoL.
Assessment of cardiovascular autonomic dysfunctionPatients with autonomic dysfunction may have an abnormally low HRV, which is indicative of parasympathetic dysfunction.34 HRV can be measured with the patient in a resting position for 10min or by HRDB, as in the NIS+7 and mNIS+7 (these two scales are pay-to-use and are again not generally applied in clinical practice). Another test permitting the evaluation of cardiac dysfunction is the above-mentioned orthostatic hypotension, which may be included in the mNIS+7 instead of HRDB.
Along with neurophysiological tests, nuclear imaging has also proven useful for assessing cardiac dysfunction. Myocardial MIBG scintigraphy uses 123-I-meta-iodobenzylguanidine (I-123 MIBG) as a tracer to evaluate cardiac sympathetic dysfunction (Table 1). To do so, I-123 MIBG is intravenously administered and heart images are acquired at different times after administration. Abnormal myocardial MIBG uptake and washout in patients with ATTRv amyloidosis is associated with cardiac sympathetic denervation due to autonomic nerve degeneration.35 The extent of MIBG imaging abnormalities can be evaluated using the late heart-to-mediastinum MIBG uptake ratio (H/M), which is calculated as the ratio between the average count per pixel in a selected heart region and that in the upper mediastinum. The lower limit of H/M in healthy individuals is estimated to be 1.6.36
Assessment of gastrointestinal autonomic dysfunctionGastrointestinal symptoms have a major impact on nutritional status, which can be evaluated by body mass index (BMI) as weight in kilograms divided by the square of height in meters. However, ATTRv amyloidosis patients with malnutrition and low levels of serum albumin may experience weight gain due to fluid retention and edema, which may result in an incorrectly normal BMI. To account for this limitation, the modified BMI (mBMI), in which the BMI is multiplied by the serum albumin level (g/L), is used to assess nutritional status in ATTRv amyloidosis patients6,12,37 (Table 1). Lower mBMI scores indicate worse nutritional status. Nutritional status in ATTRv amyloidosis is probably not only due to dysautonomia, but also other alterations such as direct gastrointestinal tract infiltration or bacterial overgrowth. Indeed, on some occasions, loss of weight may appear without any digestive symptoms, leading to the identification of occult cancer.
Assessment of urinary autonomic dysfunctionBladder dysfunction can be detected by urodynamic testing (Table 1), which uses a series of tests to assess urine holding and release.20
DiscussionAutonomic dysfunction occurs in most patients with ATTRv amyloidosis, whose highly varied symptoms can include cardiovascular manifestations, gastrointestinal disturbances, and sexual and urinary dysfunction. Autonomic manifestations usually develop at early stages of ATTRv amyloidosis, before motor impairment, due to the morphological characteristics of the vegetative nerve fibers.5 Thus, early detection of autonomic dysfunction may facilitate an earlier diagnosis of this disease. As such, autonomic-related symptoms in patients with peripheral polyneuropathy should be considered a red flag sign of ATTRv amyloidosis.1,2
Because autonomic dysfunction can be difficult to recognize due to the lack of specific symptoms or even the lack of overt symptoms, autonomic testing involving the use of different neurological and non-neurological tests is crucial for an early and accurate diagnosis. Warning signs of cardiovascular autonomic dysfunction include orthostatic hypotension and reduced HRV. Orthostatic hypotension is a common manifestation of autonomic dysfunction in ATTRv amyloidosis and may appear very early in the course of the disease. However, orthostatic variations in arterial pressure can be affected by many variables, including age, medications, and dehydration caused by diarrhea, which is another possible early symptom of autonomic dysfunction.21 Low HRV, as assessed by HRDB, has been extensively used to evaluate autonomic dysfunction, even though it reflects only an impairment of the parasympathetic component of the autonomic control of heart rate.38 Along with its limited use in patients with arrhythmias or pacemakers,23 the HRDB assessment should consider that HRV varies with age and sex.39,40 Orthostatic hypotension has been correlated with a low HRBD39 but also with noncardiac manifestations such as small fiber neuropathy with sudomotor denervation and reduced IENFD in skin biopsy.26 Another technique suitable for evaluating cardiac dysfunction is MIBG scintigraphy. This imaging technique is particularly valuable in the early detection of cardiac involvement because it may detect cardiac sympathetic denervation before symptom onset.36,41 Notably, some studies suggested that cardiac sympathetic denervation typically precedes the neurological and cardiac manifestations in ATTRv amyloidosis patients with the V30M variant,35 and it has been found even in the absence of orthostatic hypotension in non-V30M patients.11 In addition, an MIBG imaging H/M below the lower limit of normal (1.6) in ATTRv amyloidosis patients is associated with worse prognosis.36 Along with cardiac sympathetic denervation, evidence of sudomotor denervation obtained by immunostaining of skin biopsies with a neuronal marker is also a useful prognostic marker in ATTRv amyloidosis.26,36
Assessment of IENFD through skin biopsy is more specific than other methods for confirming small fiber neuropathy. However, the expertise required for its interpretation and the invasive nature of the skin biopsy make it unsuitable for widespread screening and follow-up of small fiber neuropathy. Corneal confocal microscopy has recently shown promise for small fiber testing. This noninvasive imaging technique can identify a reduced corneal nerve fiber length, which is correlated with autonomic dysfunction severity.30 In addition to the pathological evaluation of small fiber neuropathy, several neurophysiological tests can be used to detect alterations in sudomotor function. The QSART has good sensitivity, although it measures only the postganglionic sudomotor function and cannot detect preganglionic fiber damage.27 It is also a time-consuming method that requires expensive equipment and highly experienced technicians.28,29 Although the Sudoscan and SSR are both easier to perform than the QSART, the Sudoscan is more sensitive and specific than the SSR.28 Thus, the Sudoscan can be a useful tool for assessing sudomotor function due to its simplicity and accuracy.29 Importantly, a reduced electrochemical skin conductance on the Sudoscan is positively correlated with pathological evidence of sudomotor nerve fiber damage obtained through skin biopsy42,43 and negatively correlated with sensorimotor neuropathy assessed by total NIS score.44 However, the performance of the Sudoscan in ATTRv amyloidosis patients with non-V30M variants and in longitudinal studies has been poorly evaluated. Furthermore, no correlation has been found between the Sudoscan and COMPASS-31, indicating that mechanisms other than sympathetic cholinergic sudomotor dysfunction are responsible for the autonomic impairment.44
Because pathological and neurophysiological evidence of autonomic dysfunction is unfortunately difficult to obtain, self-rating questionnaires such as the COMPASS-31 and Norfolk QOL-DM have been used in clinical trials to evaluate autonomic dysfunction and QoL, respectively. Although the COMPASS-31 is a validated and quantitative measure of autonomic symptoms, it is long and complex. In addition to the COMPASS-31, brief scales such as the CADT and SAS can also be useful for assessing autonomic dysfunction. Whereas the CADT has proven efficacy and reliability in the evaluation of the main symptoms of autonomic dysfunction in ATTRv amyloidosis patients,31 the SAS needs to be further validated in ATTRv amyloidosis.
The progression of autonomic dysfunction in patients with ATTRv amyloidosis as assessed by the COMPASS-31 correlates with the sensorimotor neuropathy assessed by the NIS total score.44 For diagnostic purposes, it should be mentioned that the COMPASS-31 does not consider the sequence of symptoms and the underlying disease and the results should therefore be further verified by exploring the medical history to avoid misdiagnosis.45 Similarly, a correlation has also been demonstrated between the Norfolk QOL-DN, which includes a dysautonomic subset of questions, and the total NIS in patients with ATTRv amyloidosis.37 In clinical trials of patients undergoing treatment, improvements were observed in the COMPASS-31 score at the same time as those in the mNIS+7 and Norfolk QOL-DN total and autonomic neuropathy domain scores.6 Along with improvements in the severity of symptoms in the COMPASS-31 and Norfolk-QOL-DN, a favorable treatment effect in clinical trials was also observed with the mBMI.6,46 Thus, the mBMI may serve as an objective measure of nutritional status related to gastrointestinal manifestations, which are frequently found at early stages of the disease.
Finally, the assessment of sexual and urinary autonomic involvement should also be considered because it is another common manifestation of autonomic dysfunction in ATTRv amyloidosis that contributes to severe morbidity and to a significant decrease in QoL.
ConclusionsAutonomic dysfunction can be a warning sign of ATTRv amyloidosis and a prognostic factor of survival in patients with this disease. Although the manifestation of autonomic dysfunction primarily associated with worse survival is cardiovascular autonomic neuropathy, other factors affecting survival include gastrointestinal disturbances and baseline presence of urinary incontinence or erectile dysfunction.8 In addition, all symptoms have a significant impact on QoL.20
Given the advent of new ATTRv amyloidosis treatment options capable of slowing or even reversing disease progression,13,14,47 the early identification of autonomic disturbances is crucial to avoid diagnostic delays and the misdiagnosis of ATTRv amyloidosis. Thus, a standardized screening strategy is required for autonomic dysfunction, which should include a combination of autonomic cardiovascular assessments, pathological and neurophysiological tests, and other non-neurological tests to cover the wide variety of potential manifestations of autonomic dysfunction in patients with ATTRv amyloidosis.
Further studies addressing the correlations among different tests and their prognostic value in different pathogenic TTR variants are required to establish the minimum battery of tests for an accurate diagnosis and follow-up of autonomic dysfunction, which may help to improve the accuracy and timeliness of the diagnosis of ATTRv amyloidosis.
FundingThis study was funded by Alnylam Pharmaceuticals. Editorial support was provided by Kevin Clayton and Josep Solanes, funded by Alnylam Pharmaceuticals.
We would like to thank the group of KOLs in ATTRv amyloidosis as well as the Alnylam Medical Department for their contributions to this work: Ines Losada, PhD, MD (Internal Medicine Service, Hospital Universitario Son Llàtzer, Palma, Spain), Solange Kapetanovic García, MD (Unidad de ELA y Neuromuscular, Hospital Universitario Basurto, Bilbao, Spain), Teresa Sevilla Mantecón, MD (Hospital Universitari i Politècnic La Fe & IIS La Fe, Neuromuscular Diseases Unit, Department of Neurology, Valencia, Spain; Universitat de València, Valencia, Spain), Francisco Muñoz Beamud, MD (Internal Medicine Service, Unidad de Enfermedades Autoinmunes, Minoritarias y Trombosis, Hospital Juan Ramón Jiménez, Spain), Marta de Andrés (Alnylam Pharmaceuticals Spain S.L.) and María García-Álvaro (Alnylam Pharmaceuticals Spain S.L.).

