



Hypopituitarism is a clinical syndrome caused by insufficient secretion or a complete lack of secretion of one or several anterior pituitary hormones. Although diagnosis is typically established during the neonatal period, the initial manifestation may occasionally be a psychomotor delay in infants; therefore, early diagnosis may prevent neurocognitive impairment, thus preventing neurological sequelae.1
We present 2 cases of hypopituitarism, diagnosed after psychomotor delay was detected in infant patients.
Case 1The patient was a newborn girl born after a full-term pregnancy, who was monitored due to increased biparietal diameter and lateral ventriculomegaly with colpocephaly, and no signs of intracranial hypertension. At the age of 20 months, she had not started walking or speaking; a neurometabolic study, cytomegalovirus viral load determination, electroencephalography, auditory evoked potentials, eye fundus examination, karyotyping, and newborn screening test yielded normal results. At the age of 21 months, she was 70.3cm (−4.8 SD) long and weighed 6.9kg (−3.71 SD), displaying slow growth; results from a hormonal analysis were compatible with growth hormone (GH) deficiency, whereas the remaining studies, including a thyroid analysis, yielded normal results. The array-CGH study revealed a 1q25.2 deletion, associated with LHX4 haploinsufficiency, involved in hypopituitarism. At the age of 23 months, she showed free T4 levels of 0.51ng/dL (normal range, 0.58-1.64), TSH level of 0.97μIU/mL (normal range: 0.34-5.6), which is unusually normal given the low T4 level. All other findings for the hypothalamic-pituitary axis were normal (glycaemia, ions, ACTH, cortisol, IGF-1, IGFBP-3, and prolactin). We started treatment with oral levothyroxine (25μg/day) for central hypothyroidism, which clearly improved muscle tone and psychomotor development. A brain magnetic resonance imaging (MRI) scan revealed bilaterally increased lateral ventricular system with colpocephaly, and pituitary hypoplasia in its normal position.
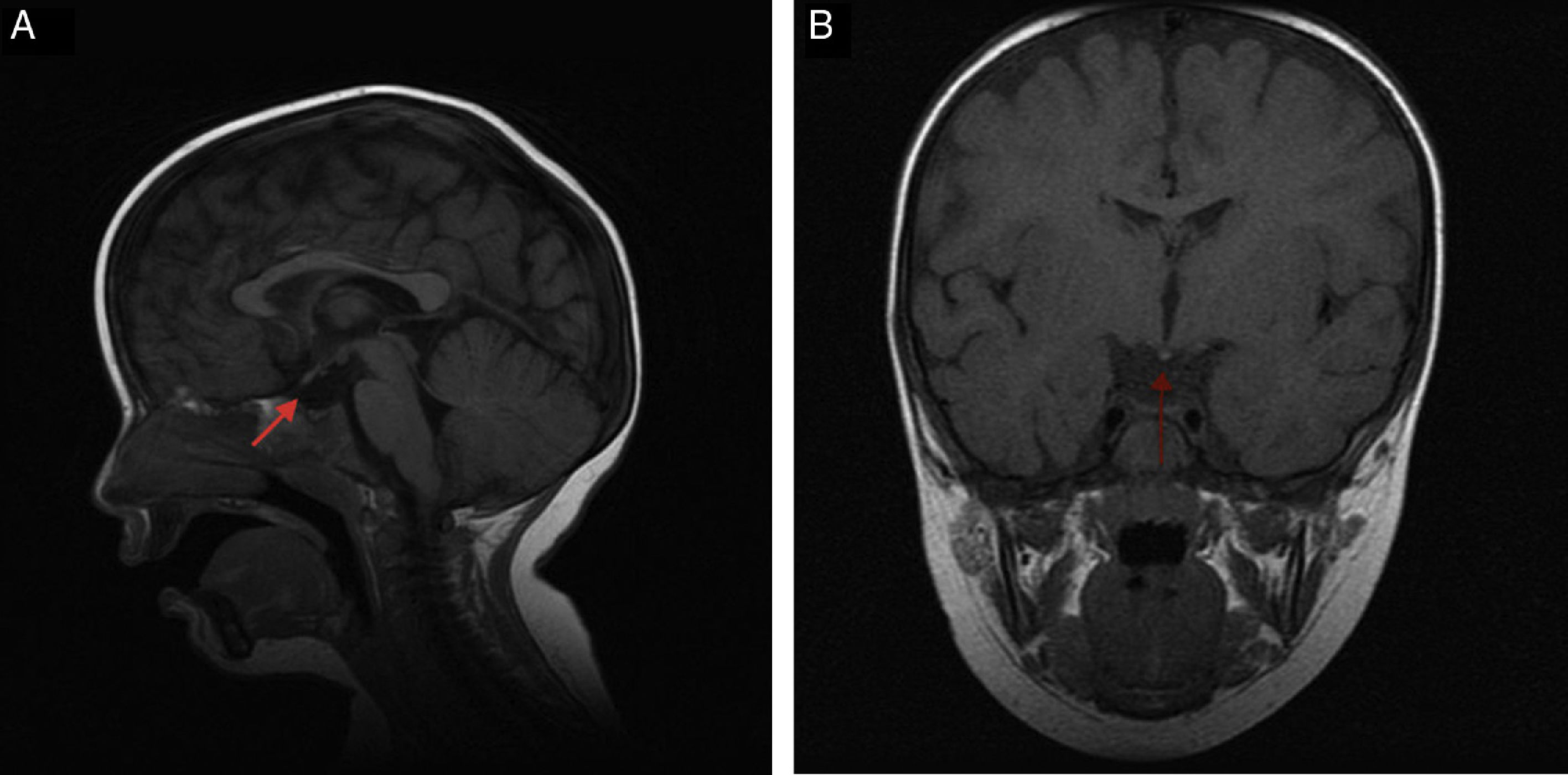
Case 2The patient was a newborn boy born after a full-term pregnancy with a history of symptomatic hypoglycaemia at birth, mucocutaneous jaundice treated with phototherapy, weak suck, and pronounced cervicoaxial hypotonia; the neonatal examination yielded normal results. At the age of 12 months, a laboratory test performed due to persistence of hypotonia, inability to sit up, and persistent constipation revealed free T4 levels of 0.50ng/dL and TSH levels of 2.34μIU/mL. All other findings for the hypothalamic-pituitary axis were normal (glycaemia, ACTH, cortisol, LH, FSH, testosterone, prolactin, IGF-1, and IGFBP-3). We started treatment with oral levothyroxine (25μg/day) for central hypothyroidism, which improved the constipation and psychomotor development. The patient presented hypoplastic scrotum, retractile testes, and penis length in the lower threshold of normality. At the age of 2, he presented significant asthenia after playing and somnolence accompanied by clumsiness and frequent falls; cortisol levels were 2.71μg/dL (normal range, 5-25); ACTH (7.7pg/mL, normal range, 5-46) and ion values were normal. We started hormone replacement therapy with hydrocortisone, which improved the symptoms. The MRI scan showed moderate anterior pituitary hypoplasia, ectopic posterior pituitary at the median eminence, and complete agenesis of the pituitary stalk (Fig. 1A and B). A genetic test yielded normal results.
 and coronal (B) slices. Moderate anterior pituitary hypoplasia, ectopic posterior pituitary at the median eminence, and complete agenesis of the pituitary stalk.")
Aetiology of hypopituitarism is varied, and includes acquired (tumours secondary to malformations of the central nervous system),2 genetic (alteration in transcription factors PIT1, PROP1, HESX1, LHX3, LHX4, PITX1, PITX2, TPIT, and SOX3),3 and idiopathic causes.
Diagnosis is clinical, as the condition presents with variable and heterogeneous symptoms, according to severity, the number of hormones affected, and how quickly symptoms manifest. The most common symptoms during the neonatal period are hypoglycaemia, prolonged jaundice with cholestasis, micropenis, midline abnormality, neurological symptoms, and psychomotor delay with hypotonia, lethargy, or weak suck.4 Suspicion of hypopituitarism is essential in order to establish a diagnosis as early as possible, thereby limiting the neurocognitive delay which is usually associated with this disorder.5
Diagnosis is established based on clinical findings and hormone level measurements (initially alterations in GH levels and the thyroid axis [TSH], followed by alterations to the adrenal [ACTH] or gonadal axis [LH/FSH], and lastly, altered prolactin levels). Diagnosis is confirmed by molecular studies or MRI, which reveals a variable lesion spectrum (small sella turcica, hypoplastic or aplastic pituitary, lack of pituitary stalk, and lack of hyperintensity in the posterior pituitary area). Treatment for hypopituitarism consists in treating the hormone deficiencies.6
In summary, early diagnosis of this syndrome prevents neurological sequelae, enabling proper neuropsychological development in later life; it is therefore very important to consider the condition in cases of psychomotor delay symptoms of unknown aetiology. It would be beneficial to include free T4 levels determination (in addition to TSH) in newborn screening, since this may enable early detection of central hypothyroidisms.
FundingThis study has received no funding of any kind.
Please cite this article as: López Úbeda M, de Arriba Muñoz A, Abenia Usón P, Labarta Aizpún JI. Hipopituitarismo. Una causa poco frecuente de retraso psicomotor. Neurología. 2018;33:551–552.
