



Childhood absence epilepsy (CAE) is considered easily manageable with medication provided that a strict patient classification system is employed. It accounts for 10% of all childhood epilepsy cases starting before the age of 15 and it is most frequent in school-aged girls. The aim of this study is to analyse long-term outcomes of patients diagnosed with CAE according to the Loiseau and Panayiotopoulos criteria and treated during childhood.
MethodsWe conducted a retrospective study including 69 patients with CAE who are currently older than 11; data were gathered from medical histories, EEG records, and telephone questionnaires.
Results52 patients met the Loiseau and Panayiotopoulos criteria. Mean age is now 17.16 years. Female-to-male ratio was 1.65:1; mean age at onset was 6 years and 2 months; mean duration of treatment was 3 years and 9 months. A family history of epilepsy was present in 30.8% of the patients and 7.7% had a personal history of febrile convulsions. Absence seizures were simple in 73.5% of the patients and complex in 26.5%. Response rates to first-line treatment were as follows: valproic acid, 46.3%; and valproic acid plus ethosuximide, 90.9%. The rate of response to second-line therapy (ethosuximide or lamotrigine) was 84.2%; 4% of the patients experienced further seizures after treatment discontinuation, 78.8% achieved seizure remission, and 25% needed psychological and academic support.
ConclusionsOur data show that epileptic patients should be classified according to strict diagnostic criteria since patients with true CAE have an excellent prognosis. The relapse rate was very low in our sample. Despite the favourable prognosis, psychological and academic support is usually necessary.
La epilepsia ausencia infantil (EAI) se considera una forma de epilepsia de fácil control farmacológico solo si se emplean criterios estrictos para la clasificación de los pacientes. Supone el 10% de las epilepsias infantiles de inicio antes de los 15 años y es más frecuente en niñas escolares. El objetivo es conocer la evolución a largo plazo de los pacientes atendidos en la etapa infantil con EAI empleando los criterios de Loiseau y Panayiotopoulos.
MétodosEstudio retrospectivo de 69 pacientes con EAI con edad actual mayor de 11 años, realizado mediante revisión de historias clínicas, EEG y cuestionario telefónico.
ResultadosCumplieron los criterios de Loiseau y Panayiotopoulos 52 pacientes, edad actual media 17,61 años. Relación mujeres/hombres: 1,65/1; edad de inicio media: 6 años y 2 meses; duración total de tratamiento media: 3 años y 9 meses; antecedentes familiares de epilepsia: 30,8%; antecedentes personales de crisis febriles: 7,7%; tipo de ausencias: simples 73,5%, complejas: 26,5%; respuesta al primer tratamiento: ácido valproico 46,3% o ácido valproico con etosuximida simultáneos 90,9%; respuesta al segundo tratamiento (etosuximida o lamotrigina) 84,2%; crisis tras supresión de tratamiento: 4%; pacientes en remisión terminal: 78,8%; necesidad de apoyo psicopedagógico: 25%.
ConclusionesNuestros datos muestran la utilidad de clasificar a los pacientes utilizando criterios estrictos ya que el pronóstico de las crisis del síndrome de EAI puro es excelente. Encontramos que la tasa de recaídas ha sido muy baja. A pesar del favorable pronóstico en cuanto al control de crisis necesitan apoyos psicopedagógicos en un alto porcentaje.
Absence seizures are a type of epileptic seizure defined by Wyllie et al.1 as a sudden-onset seizure interrupting ongoing activities. During the seizure, the patient shows distant gaze, sometimes with upgaze and blinking; if the patient is speaking, speech is slowed or interrupted; if walking, he or she stands transfixed; if eating, he or she will stop the food on the way to the mouth. The patient will be unresponsive when spoken to. In some, attacks are aborted when the patient is spoken to. The attack lasts from a few seconds to half a minute and evaporates as rapidly as it commenced.1 Absence seizures may be classified as typical or atypical according to their electroclinical characteristics. Typical absence seizures involve suppression of mental functions, including comprehension, reactivity, and memory; they characteristically start and end abruptly and usually last 5-15seconds. Ictal EEG shows generalised synchronous and symmetrical 3-Hz spike-and-wave discharges.2 Typical absence seizures are included in numerous epileptic syndromes listed in the International League Against Epilepsy classification of epilepsies and epileptic syndromes, such as childhood absence epilepsy (CAE), the subject of our study, which is included in the group of idiopathic generalised epilepsies.3
CAE is characterised by typical absence seizures, both simple and complex, as the only type of seizure at onset, with multiple seizures per day, and onset before puberty with normal psychomotor development and ictal EEG abnormalities corresponding to regular, bilateral, symmetrical, and synchronous 3-Hz spike-and-wave discharges with normal or slightly altered background activity. This type of generalised epilepsy is frequent in children aged 6-10 years,4 representing 10%-17% of all epilepsies diagnosed in school-age children,5 with a prevalence rate of 1.5%-12% depending on the series6; CAE is more frequent in girls, with some exceptions. Regarding prognosis, the published remission rates vary according to the diagnostic criteria used. According to the latest criteria by Panayiotopoulos,7 revised by Loiseau et al.,8 which are stricter than those proposed by the International League Against Epilepsy in 1989, the remission rate may reach 90%. These criteria lead to a more homogeneous group of patients and enable better understanding of CAE progression.9 This epileptic syndrome has classically been assigned to the group of “benign” epilepsies due to the high rate of seizure control, although we now know that up to 38% of patients with CAE will suffer psychosocial, academic, or professional problems,10 and up to 35% may develop significant attention difficulties despite having a normal intelligence quotient.11
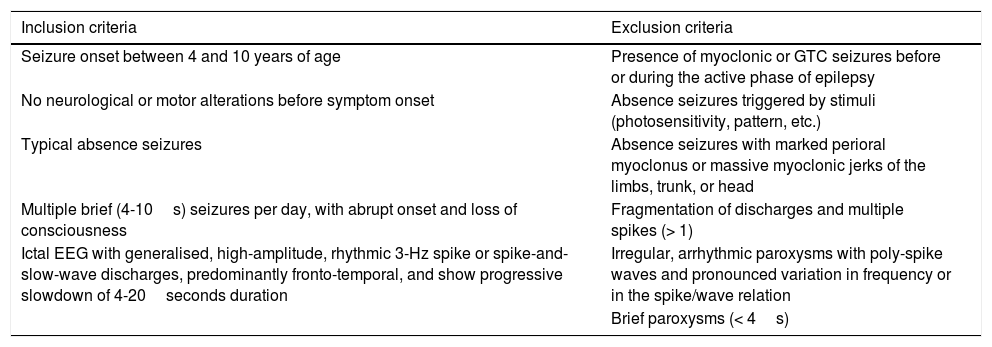
The aim of this study is to analyse the long-term progression of paediatric patients with CAE, who were selected using the Loiseau and Panayiotopoulos criteria (Table 1).
International League Against Epilepsy (1989) and Loiseau and Panayiotopoulos (2005) criteria (adapted for this study).
| Inclusion criteria | Exclusion criteria |
|---|---|
| Seizure onset between 4 and 10 years of age | Presence of myoclonic or GTC seizures before or during the active phase of epilepsy |
| No neurological or motor alterations before symptom onset | Absence seizures triggered by stimuli (photosensitivity, pattern, etc.) |
| Typical absence seizures | Absence seizures with marked perioral myoclonus or massive myoclonic jerks of the limbs, trunk, or head |
| Multiple brief (4-10s) seizures per day, with abrupt onset and loss of consciousness | Fragmentation of discharges and multiple spikes (> 1) |
| Ictal EEG with generalised, high-amplitude, rhythmic 3-Hz spike or spike-and-slow-wave discharges, predominantly fronto-temporal, and show progressive slowdown of 4-20seconds duration | Irregular, arrhythmic paroxysms with poly-spike waves and pronounced variation in frequency or in the spike/wave relation |
| Brief paroxysms (< 4s) |
EEG: electroencephalography; GTC: generalised tonic–clonic seizures.
We performed a retrospective study of patients diagnosed with CAE between 1988 and 2015 and aged older than 11 at the time of study inclusion. Patients were drawn from 2 paediatric neurology consultations (one at a hospital and the other outside hospital) attending patients with similar demographic and clinical characteristics and using homogeneous diagnostic and treatment criteria. We gathered the following data from patients’ medical records: sex, family history of epilepsy, history of febrile convulsions, type and age of onset of absence seizures, time of progression to diagnosis, drug use and response, EEG at diagnosis, progression, and treatment duration. Furthermore, we administered a voluntary, structured telephone questionnaire on seizure recurrence after treatment discontinuation, type of seizures, terminal remission (in our study, one year with no treatment and no seizures), current treatment, need for psycho-paedagogical support, and psychological problems.
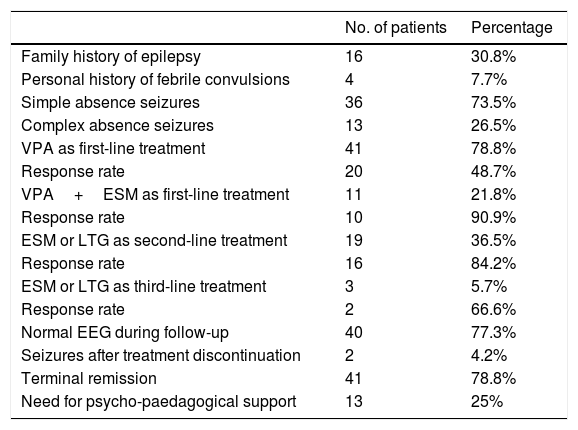
ResultsWe identified 69 patients diagnosed with CAE between 1988 and 2015 and older than 11 at the time the present study was conducted. After applying the Loiseau and Panayiotopoulos criteria, we excluded 17 patients; the total number of patients included was therefore 52. Five patients could not be located, so the telephone questionnaire on long-term outcomes was administered to 47 (90%). Mean age was 17.61 years (SD 5.79; range, 11-36); mean age at onset of absence seizures was 6 years (73.8 months, SD 21.77; range, 4 years to 9 years and 6 months). Of all patients, 32 (62%) were female and 20 male (38%); the female-to-male ratio was 1.65:1.
Sixteen patients (30.8%) had a family history of epilepsy and 4 (7.7%) a personal history of febrile convulsions. Regarding the type of absence, 36 patients (73.5%) presented simple absence seizures and 13 (26.5%) complex absence seizures. Progression time from symptom onset to the first consultation was 6 months (25.63 weeks, SD 24.12) and mean treatment duration was 3 years and 9 months (45.31 months; SD 31.01).
The initial treatment was valproic acid (VPA) in monotherapy in 41 patients; of these, 48% remained seizure-free. Eleven patients received a combination of VPA and ethosuximide (ESM) from onset, with a response rate of 90.9%. After the first-line treatment, 19 patients continued presenting seizures; 16 received ESM and 3 lamotrigine (LTG), with 16 achieving seizure control (response rate of 84.2%). The remaining 3 patients needed a third drug (ESM or LTG); 2 (66%) responded to this treatment. Mean treatment duration was 3.5 years (maximum of 7 years). During follow-up, EEG abnormalities persisted in 22.7% of patients. Only 2 patients (4%) presented seizures after treatment discontinuation, one patient presented absence seizures and the other myoclonic seizures. Terminal remission was achieved in 78.8% of patients. Twenty-five percent of patients needed psycho-paedagogical support (Table 2).
Results.
| No. of patients | Percentage | |
|---|---|---|
| Family history of epilepsy | 16 | 30.8% |
| Personal history of febrile convulsions | 4 | 7.7% |
| Simple absence seizures | 36 | 73.5% |
| Complex absence seizures | 13 | 26.5% |
| VPA as first-line treatment | 41 | 78.8% |
| Response rate | 20 | 48.7% |
| VPA+ESM as first-line treatment | 11 | 21.8% |
| Response rate | 10 | 90.9% |
| ESM or LTG as second-line treatment | 19 | 36.5% |
| Response rate | 16 | 84.2% |
| ESM or LTG as third-line treatment | 3 | 5.7% |
| Response rate | 2 | 66.6% |
| Normal EEG during follow-up | 40 | 77.3% |
| Seizures after treatment discontinuation | 2 | 4.2% |
| Terminal remission | 41 | 78.8% |
| Need for psycho-paedagogical support | 13 | 25% |
EEG: electroencephalography; ESM: ethosuximide; LTG: lamotrigine; VPA: valproic acid.
Multiple studies analyse long-term progression of CAE but use different classification and methodological criteria; therefore, the data available in the literature are heterogeneous. In light of this evidence, we decided to use strict criteria in our study,7,8 specifically the Loiseau and Panayiotopoulos criteria, to ensure a homogeneous sample despite patients being drawn from 2 different consultations. We present a series of patients with “pure” CAE, which is larger than other series published in our setting.4,12 The retrospective design of our study entails the limitations inherent to this type of study: bias in data collection and memory bias, which may affect the results.
CAE generally manifests between 4 and 10 years of age, peaking between 5 and 7 years,13 and is clearly more frequent in girls, with some exceptions.14 These characteristics were also observed in our series: patients had a mean age of 73.8 months (6 years) and were mainly female (62% were girls), which characterises CAE as a frequent epilepsy among school-age girls. The mean age of 17.61 years at the time of study inclusion is the result of the long follow-up period. The frequency of family history of epilepsy (30.8%) and personal history of febrile convulsions (8%) observed is consistent with published data, which reflects a predisposing genetic basis.
In our study, 73.5% of patients presented simple absence seizures and 26.5% complex absence seizures, similar to the results reported in the literature. Mean progression time from symptom onset to the first consultation was more than 6 months (25.63 weeks), probably due to the subtle character of the seizures, which frequently go unnoticed or are misdiagnosed as tics, distractions, or sterotyped movements.
The drugs most frequently used to treat CAE are VPA, ESM, and LTG; however, only 6 trials of these drugs in monotherapy were performed before 2010, with limited numbers of patients; considering the methodological limitations, none showed sufficient evidence to offer robust recommendations in clinical practice.15 In 2010, a randomised, double-blind, placebo-controlled, multicentre study of 453 patients was conducted to compare the efficacy, tolerability, and adverse effects of VPA, ESM, and LTG. This study showed that VPA is not more effective than ESM or LTG and causes more cognitive adverse reactions than ESM.16 Studies of rats with 2 different genetic models of CAE show that ESM not only controls seizures but may also have a “disease-modifying role.”17,18 Similarly, a 2014 prospective study assessing the long-term progression of patients according to the initial treatment used (VPA or ESM) revealed a clear tendency to maintain a higher rate of full remission when patients were treated with ESM and not with VPA, regardless of EEG abnormalities.19 In our series, VPA was used as the first-line treatment, either in monotherapy or in association with ESM, in all patients. The effectiveness of VPA was modest compared with the data available in the literature (48.7% in our series compared to 77%-85% in the literature),12,20 probably because we combined VPA and ESM where there was no initial response, without increasing VPA doses to those reported in other studies. Interestingly, patients whose initial treatment was combination therapy (VPA+ESM) achieved a seizure control rate of 90% with no further adverse effects. Our literature review identified no similar studies, hence we cannot compare these results.
There are no universally accepted criteria on treatment duration; a period of between one and 2 years without seizures and normal EEG readings is generally recommended to start discontinuation.15 We observed higher mean duration in our patients (3.5 years), with cases of up to 7 years, which indicates that, despite the good prognosis of CAE, long-term follow-up is necessary, especially in cases with an initially poor response.
Relapses with absence seizures after treatment discontinuation occur in 8% of patients with “pure” CAE.9 The 4% observed in our study may be influenced by the retrospective character of the study or by the longer treatment duration.
The percentage of young patients developing tonic-clonic seizures has been established at 35%-60% according to various studies21; onset usually occurs 5-10 years after onset of absence seizures.22 We excluded patients with generalised tonic-clonic seizures occurring before terminal remission.
The terminal remission rate for this type of epilepsy is high, but varies according to the methodology and the concept of terminal remission used in each study; rates of 50%-90% have been published. We defined terminal remission as a period of one year with no seizures or treatment; compared with similar retrospective studies, the 78.8% of patients with terminal remission in our series coincides with the figure published in 2005 by Grosso et al.9
The good prognosis of CAE is well known; however, like other authors,10,11 we observed a very high percentage of patients with psycho-paedagogical problems (25% in our case). According to other series, up to 38% of patients with CAE will experience psychosocial, academic, or professional problems.10 Up to 35% of patients may present significant attention difficulties despite having a normal intelligence quotient.11 These attention problems interfere with memory and executive functions, and therefore with academic performance; however, patients do not detect this lack of attention in most cases. Furthermore, studies with small samples have detected visuospatial, verbal learning, memory,23,24 and language difficulties.25
We may conclude that CAE diagnosed according to strict criteria such as those proposed by Loiseau and Panayiotopoulos presents a favourable prognosis. In some patients in our series, treatment was prolonged more than stipulated in classic recommendations, probably to prevent relapses. We consider that ESM may be offered as the first-line treatment for CAE diagnosed according to strict criteria. We underscore the significant percentage of psycho-paedagogical difficulties that persist despite seizure control and antiepileptic treatment discontinuation.
Conflicts of interestThe authors have no conflicts of interests to declare.
Please cite this article as: Martínez-Ferrández C, Martínez-Salcedo E, Casas-Fernández C, Alarcón-Martínez H, Ibáñez-Micó S, Domingo-Jiménez R. Epilepsia ausencia infantil. Pronóstico a largo plazo. Neurología. 2019;34:224–228.
