





Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease among adults worldwide. It is characterised by the death of dopaminergic neurons in the substantia nigra pars compacta and, in some cases, presence of intracytoplasmic inclusions of α-synuclein, called Lewy bodies, a pathognomonic sign of the disease. Clinical diagnosis of PD is based on the presence of motor alterations. The treatments currently available have no neuroprotective effect. The exact causes of PD are poorly understood. Therefore, more precise preclinical models have been developed in recent years that use induced pluripotent stem cells (iPSC). In vitro studies can provide new information on PD pathogenesis and may help to identify new therapeutic targets or to develop new drugs.
La enfermedad de Parkinson (EP) es la segunda enfermedad neurodegenerativa más común a nivel mundial en adultos mayores. Se caracteriza por la pérdida de neuronas dopaminérgicas (nDAs) en la sustancia nigra pars compacta del mesencéfalo y en algunos casos acompañada de la aparición de cuerpos intracitoplásmaticos de Lewy de α-sinucleína, signo patognomónico de la enfermedad. La EP se diagnostica clínicamente por la presencia de alteraciones motoras principalmente y en la actualidad los tratamientos presentan nula actividad neuroprotectora. Aún no se han establecido las causas exactas de la EP, por lo que, en los últimos años se ha buscado el desarrollo de modelos preclínicos más precisos, utilizando células troncales pluripotentes inducidas (iPSCs). Permitiendo el estudio de la enfermedad de manera in vitro para generar conocimiento novedoso sobre su patogénesis y el descubrimiento de nuevos posibles blancos terapéuticos o el desarrollo de nuevos fármacos.
Parkinson’s disease (PD) is the neurodegenerative disease with the second highest incidence rate globally, and presents during adulthood in the majority of cases.1 The disease is characterised by 2 main pathological processes: loss of dopaminergic neurons (DN) in the substantia nigra pars compacta (SNpc) of the ventral midbrain,2 and the presence of intracellular aggregates of α-synuclein protein, known as Lewy bodies, in the same region.3 Clinically, diagnosis is based on 4 distinctive motor alterations: resting tremor, muscle rigidity, postural instability, and bradykinesia.4 A series of non-motor alterations have recently been associated with the disease; these include cognitive impairment, depression, sleep alterations, and loss of the sense of smell.5
Currently, only symptomatic treatments are available, and no neuroprotective drug has been identified.6 The most frequently used drug, levodopa, has been in use since the 1960s to control the motor symptoms of PD.7,8 Levodopa is an amino acid that stimulates dopamine (DA) receptors through the action of the DOPA decarboxylase enzyme in the brain.9 Stem cells can be used as a preclinical model to study such neurodegenerative diseases as PD in vitro: they present high proliferative capacity, are able to mimic different stages of the disease, and are easier to obtain than the post mortem tissue samples that are often used to study this type of disease.10
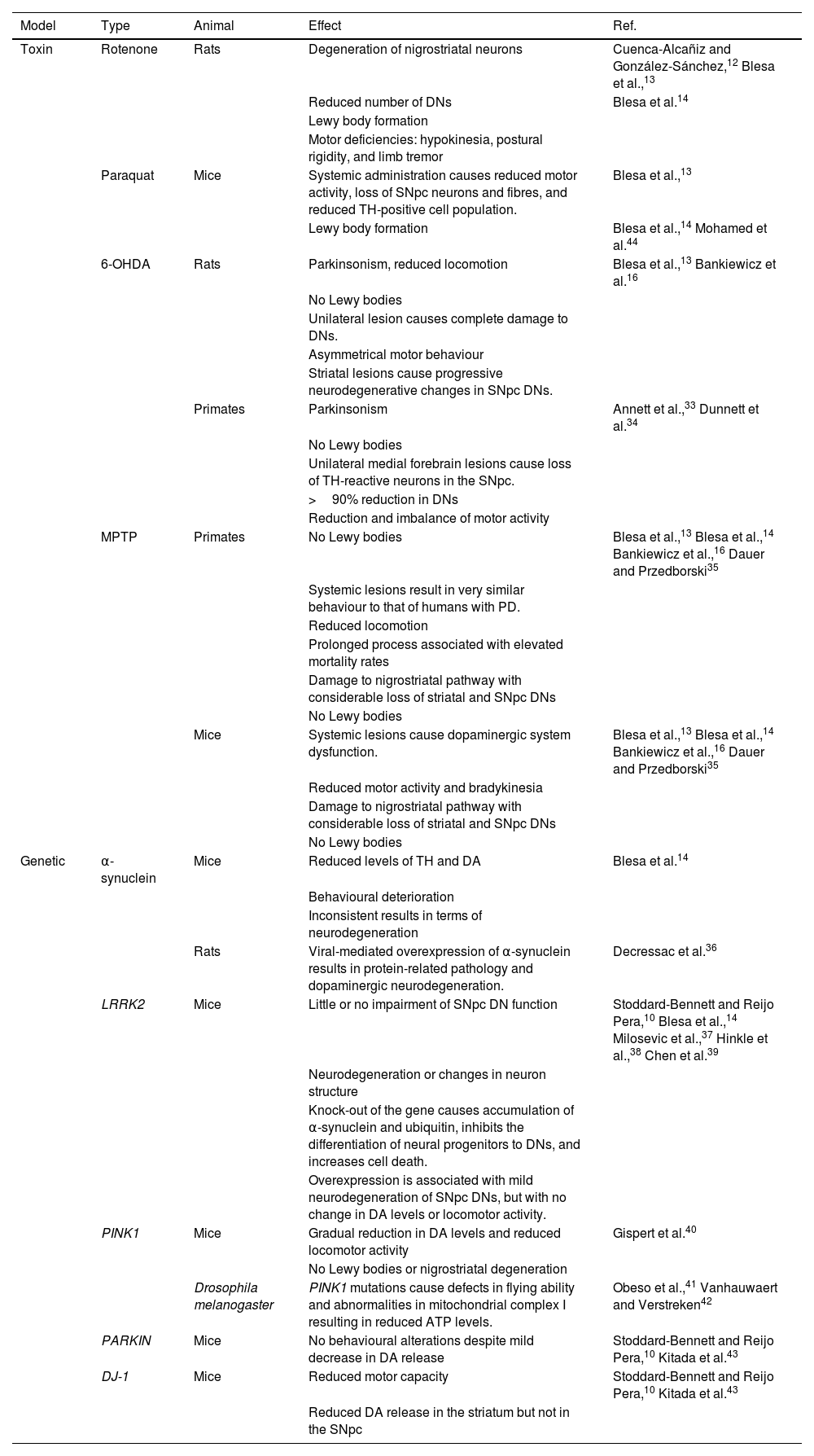
Animal models used in Parkinson’s diseaseNumerous animal models have been developed to mimic the neuropathological lesion occurring in PD. However, none of these is able to reproduce the human disease; for this reason, a series of models and techniques are used to study different aspects of PD in human patients, such as the high sensitivity of DNs, the formation of Lewy bodies, and movement alterations.10,11 Two types of animal model have been developed: neurotoxin-induced disease and genetic modification. The most widely studied neurotoxins are rotenone, paraquat, 6-hydroxydopamine (6-OHDA), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).12–14 A third model is based on partial blockade of the nigrostriatal DA pathway secondary to a mechanical lesion to the pathway at the medial forebrain bundle, resulting in progressive degeneration of SNpc DNs, mimicking PD.15
The most frequently used animals in PD models are rats, mice, zebrafish, Drosophila melanogaster fruitflies, the nematode Caenorhabditis elegans, and non-human primates.16D. melanogaster PD models show reduced locomotion and difficulty flying.17C. elegans displays a reduced basal slowing response, shorter survival, and alterations in defecation and reproduction cycles, which constitute phenotypic characteristics of PD.18,19 Zebrafish models present alterations in locomotor activity, with reduced crosses and swimming distance and speed, as well as increased number and duration of freezing episodes.20,21 Murine models enable analysis of the disease from an anatomical, biochemical, and behavioural perspective, offering simple management and high reproducibility in a model reflecting late stages of PD.22,23 Studies of behavioural changes in mice show reduced coordination, balance, gastrointestinal function, stride length, and olfactory acuity; difficulty in nest building, and impaired ability to walk.24,25 Rat models usually display limb rigidity, cognitive deficits, reduced motor activity, rotational behaviours, hypokinesia, bradykinesia, and postural asymmetry.20,26 Non-human primates show behavioural changes analogous to those observed in patients with PD, including bradykinesia, limb rigidity, postural disturbances, difficulty balancing, resting tremor, stable bilateral parkinsonian syndrome, gesture instability, and impaired gross and fine motor skills.16,20,27
Neurotoxin modelsOne of the most widely used neurotoxin models involves administration of rotenone, a fat-soluble pesticide and insecticide that causes oxidative stress, selectively damaging DNs via inhibition of complex I of the mitochondrial respiratory chain, resulting in the characteristic motor deficiencies of PD.12,13,28 This mouse PD model reproduces the behavioural alterations observed in humans, and presents intracellular inclusions resembling Lewy bodies.14,29 In rat models, exposure to rotenone causes degeneration of DNs and the formation of intracellular inclusions similar to Lewy bodies. These effects result in motor deficiencies similar to those occurring in PD, including hypokinesia, postural rigidity (stooped posture), and limb tremor.12,13
Another toxin used in rat models is paraquat (1,1'-dimethyl-4-4'-bipyridinium dichloride), commonly used as a herbicide, which produces free radicals that react with the cellular lipid membrane. The compound shows a certain selectivity for tyrosine hydroxylase (TH)–positive SNpc DNs.12,13,30 Systemic administration of paraquat in rats results in a reduction in motor activity, with a decrease in numbers of TH-positive neurons and fibres in the SNpc, and can lead to the development of Lewy bodies; however, variable results are reported with regard to neuron death.14
To date, the most widely used toxin has been 6-OHDA, whose metabolism leads to the formation of free radicals, inhibiting the mitochondrial respiratory chain. The toxin presents 3 important characteristics: 1) it induces rapid degeneration; 2) it displays great affinity for norepinephrine and DA transporters, causing death of adrenergic and dopaminergic neurons; and 3) as it does not cross the blood-brain barrier, systemic administration of the toxin does not induce parkinsonism, and direct intracerebral injection is required.12–14,31,32 In non-human primates, a unilateral lesion to the medial forebrain causes loss of TH-immunoreactive neurons in the SNpc and loss of over 90% of DNs, resulting in a reduction and imbalance of motor activity.33,34
In rats, unilateral 6-OHDA lesions cause complete damage in DNs, resulting in asymmetrical motor activity; thus, this is an ideal model for studying cell replacement therapies and neuroprotective factors. Furthermore, partial lesions require a reduction of the doses used in unilateral lesions; the striatal lesion model is used to study pathophysiological and neurodegenerative mechanisms, as these lesions cause progressive neurodegenerative changes in SNpc DNs.16
The fourth most popular toxin used in PD models is MPTP, a protoxin whose metabolism by monoamine oxidase B produces the metabolite 1-methyl-4-phenyl pyridinium (MPP+). Its action mechanism is based on excessive release of DA, whose metabolism results in excessive generation of reactive oxygen species and free radicals. MPP+ also inhibits complex I of the mitochondrial electron transport chain, reducing the production of adenosine triphosphate. However, this toxin does not selectively damage SNpc DNs, and usually does not induce the formation of Lewy bodies.12,13
MPTP models in non-human primates contribute information about potential treatments and pathogenic mechanisms of PD: systemic lesions cause very similar behaviour to that observed in human patients with PD. However, this process is prolonged and is associated with an elevated mortality rate. In contrast, mice present neuropathological and biochemical characteristics of DA system damage, in addition to reduced motor activity. In mouse models, systemic lesions cause a degree of impairment to the DA system; this is ideal for studying pathophysiological and neurodegenerative processes.16 In both species, MPTP damages the nigrostriatal pathway, with significant loss of striatal and SNpc DNs; the major disadvantage of these models is that Lewy bodies are not observed (Table 1).14,35
Comparison of different animal models of Parkinson’s disease and their respective advantages and disadvantages.
| Model | Type | Animal | Effect | Ref. |
|---|---|---|---|---|
| Toxin | Rotenone | Rats | Degeneration of nigrostriatal neurons | Cuenca-Alcañiz and González-Sánchez,12 Blesa et al.,13 |
| Reduced number of DNs | Blesa et al.14 | |||
| Lewy body formation | ||||
| Motor deficiencies: hypokinesia, postural rigidity, and limb tremor | ||||
| Paraquat | Mice | Systemic administration causes reduced motor activity, loss of SNpc neurons and fibres, and reduced TH-positive cell population. | Blesa et al.,13 | |
| Lewy body formation | Blesa et al.,14 Mohamed et al.44 | |||
| 6-OHDA | Rats | Parkinsonism, reduced locomotion | Blesa et al.,13 Bankiewicz et al.16 | |
| No Lewy bodies | ||||
| Unilateral lesion causes complete damage to DNs. | ||||
| Asymmetrical motor behaviour | ||||
| Striatal lesions cause progressive neurodegenerative changes in SNpc DNs. | ||||
| Primates | Parkinsonism | Annett et al.,33 Dunnett et al.34 | ||
| No Lewy bodies | ||||
| Unilateral medial forebrain lesions cause loss of TH-reactive neurons in the SNpc. | ||||
| >90% reduction in DNs | ||||
| Reduction and imbalance of motor activity | ||||
| MPTP | Primates | No Lewy bodies | Blesa et al.,13 Blesa et al.,14 Bankiewicz et al.,16 Dauer and Przedborski35 | |
| Systemic lesions result in very similar behaviour to that of humans with PD. | ||||
| Reduced locomotion | ||||
| Prolonged process associated with elevated mortality rates | ||||
| Damage to nigrostriatal pathway with considerable loss of striatal and SNpc DNs | ||||
| No Lewy bodies | ||||
| Mice | Systemic lesions cause dopaminergic system dysfunction. | Blesa et al.,13 Blesa et al.,14 Bankiewicz et al.,16 Dauer and Przedborski35 | ||
| Reduced motor activity and bradykinesia | ||||
| Damage to nigrostriatal pathway with considerable loss of striatal and SNpc DNs | ||||
| No Lewy bodies | ||||
| Genetic | α-synuclein | Mice | Reduced levels of TH and DA | Blesa et al.14 |
| Behavioural deterioration | ||||
| Inconsistent results in terms of neurodegeneration | ||||
| Rats | Viral-mediated overexpression of α-synuclein results in protein-related pathology and dopaminergic neurodegeneration. | Decressac et al.36 | ||
| LRRK2 | Mice | Little or no impairment of SNpc DN function | Stoddard-Bennett and Reijo Pera,10 Blesa et al.,14 Milosevic et al.,37 Hinkle et al.,38 Chen et al.39 | |
| Neurodegeneration or changes in neuron structure | ||||
| Knock-out of the gene causes accumulation of α-synuclein and ubiquitin, inhibits the differentiation of neural progenitors to DNs, and increases cell death. | ||||
| Overexpression is associated with mild neurodegeneration of SNpc DNs, but with no change in DA levels or locomotor activity. | ||||
| PINK1 | Mice | Gradual reduction in DA levels and reduced locomotor activity | Gispert et al.40 | |
| No Lewy bodies or nigrostriatal degeneration | ||||
| Drosophila melanogaster | PINK1 mutations cause defects in flying ability and abnormalities in mitochondrial complex I resulting in reduced ATP levels. | Obeso et al.,41 Vanhauwaert and Verstreken42 | ||
| PARKIN | Mice | No behavioural alterations despite mild decrease in DA release | Stoddard-Bennett and Reijo Pera,10 Kitada et al.43 | |
| DJ-1 | Mice | Reduced motor capacity | Stoddard-Bennett and Reijo Pera,10 Kitada et al.43 | |
| Reduced DA release in the striatum but not in the SNpc |
6-OHDA: 6-hydroxydopamine; ATP: adenosine triphosphate; DA: dopamine; DN: dopaminergic neuron; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; PD: Parkinson’s disease; SNpc: substantia nigra pars compacta; TH: tyrosine hydroxylase.
Three main brain areas are targeted in neurotoxin models of PD: the striatum, medial forebrain bundle, and substantia nigra.11 Damage to these brain areas activates compensatory mechanisms that seek to maintain neurological activity by modifying DA synthesis and release, increasing TH activity, and modifying activity in the striatum, cerebellum, and cortical areas; this has an impact on clinical aspects of PD.23
Genetic modelsGenetic models mimic the mechanisms involved in genetic forms of PD. It should be noted that PD is only genetic in 5% to 10% of cases.4 In genetic forms, the pathological and behavioural phenotypes reported in murine models tend to differ from those observed in human patients, mainly in studies of the SNCA, LRRK2, PINK1, PARKIN, and DJ-1 genes.14 These studies follow 3 main approaches: knock-out, overexpression, and transgenes. However, experimental models with D. melanogaster, C. elegans, and murine species do not display the typical motor deficiencies observed in humans; rather, studies using these models focus on the genetic form of PD, studying specific genes related to the disease.23
The SNCA gene encodes α-synuclein, the main component of the Lewy bodies observed in PD. Transgenic mice present reduced levels of TH and DA, with behavioural repercussions.14 Rat models using viral-mediated α-synuclein overexpression have reported disease related to the protein and dopaminergic neurodegeneration; as a result, this represents an ideal model for testing new neuroprotective strategies.36
LRRK2 is required for neuron survival, and is the most common target in studies using gene editing to create models of genetic PD.10 In mouse models, little or no effect on SNpc DNs is observed.14 However, knock-out mouse models have achieved α-synuclein accumulation, inhibition of the differentiation of neural progenitor cells to DNs, and increased cell death.10,37,38 Overexpression of the gene is associated with mild degeneration of SNpc DNs, but with no change in DA levels or locomotor activity.39
Models with mice lacking the PINK1 gene, essential to neuron survival under oxidative stress,10 have shown a gradual reduction in DA levels and reduced locomotor activity, without Lewy bodies or nigrostriatal degeneration.40 Studies of PINK1 mutations in D. melanogaster report defects in flying ability and abnormalities in mitochondrial complex I.41,42
Studies with mice lacking the PARKIN gene have found no behavioural changes, despite the mild decrease in DA release. Finally, models using mice with mutations in the DJ-1 gene, essential in resistance against oxidative stress, have shown decreased motor capacity and reduced DA release in the striatum but not in the SNpc (Table 1).10,43
Preclinical models for cell therapyCurrently, the most widely used treatments are pharmacological, based on DA replacement or administration of DA agonists; however, these have the disadvantage that their effectiveness is reduced as the disease advances, and can cause various adverse reactions.45,46 Surgical treatment and deep brain stimulation can lead to haemorrhage, infections, and neuropsychiatric adverse effects.47 The use of stem cells to generate DNs for transplantation represents a great advance in the future of cell therapy for such diseases as PD, aiming to achieve survival of the engrafted cells, which would form connections with the patient’s brain, leading to measurable clinical improvements.48,49 However, clinical application of these new approaches first requires in vitro and in vivo preclinical models and standardisation of critical factors (eg, patient selection, graft placement, cellular composition of the graft, and immunological regulation) to ensure the efficacy and safety of the procedure.48,50,51
Source tissues used for the acquisition of DNs include human fetal ventral mesencephalic (hfVM) tissue, the adrenal medulla, olfactory bulb, carotid body, embryonic stem cells, neural stem cells, mesenchymal stem cells, induced pluripotent stem cells (iPSC), and human parthenogenetic stem cells.45,46
Adrenal medulla transplants were the first to be studied for the treatment of PD.46 In a clinical study in humans, tissues were transplanted from the adrenal medulla, hfVM, and fetal adrenal gland. Adrenal medulla transplantation was associated with symmetrical bilateral improvements, with reduced stiffness, postural instability, and gait alterations.52,53 Tissue from the hfVM considerably improved stiffness, postural instability, gait alterations, bradykinesia, and facial expression, although tremor persisted. Fetal adrenal tissue improved stiffness and bradykinesia only.54 However, this line of research has been discontinued due to the high mortality rates associated with abdominal and cranial surgical procedures.46 In addition to the development of other treatment methods, such as transplantation of hfVM tissue, whose cells may differentiate to DNs, these studies demonstrated good histological and functional recovery, with no tumour formation in mice and non-human primates.55 A clinical study in which human patients underwent bilateral transplantation in the putamen demonstrated graft survival despite the advance of PD and continued pharmacological treatment: the striatum was reinnervated and controlled DA release was re-established, with integration into the nigrostriatal circuit.48 In turn, this approach presents surgical complications and is hindered by the technical difficulty of dissecting fetal tissue, resulting in a combination of cell populations with high mortality rates, in addition to the limited availability of fetal tissue, ethical issues, and the risk of postoperative dyskinesia.56,57
Neural stem cells derived from adults present the same properties as neural progenitor cells from the fetal nervous system. However, their behaviour is determined by the extracellular environment in which they reside.46,58 Due to the contact with the endothelial cells of blood vessels, they constitute the neurovascular niche, and release factors promoting their proliferation and genesis.59 In turn, astrocyte-like cells near the olfactory bulb have the capacity to self-renew and give rise to neuroblast progenitor cells, although their capacity to form functional DNs has not been established.46,55 One study assessed whether olfactory ensheathing glial cells allow continuous re-entry of axon fibres into the olfactory bulb during adulthood,60 demonstrating that transplantation of a combination of peripheral and central nerve grafts provides a scaffold promoting axonal growth and DA innervation in the striatum.61 Another type of neural stem cell has been described in the carotid body,46 a chemoreceptor organ derived from the neural crest that is composed of neuronal glomus cells ensheathed in glial-like cells.62 The neuronal cells contain vesicles storing high levels of DA, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor, suggesting they play a role in neurogenesis, neuroprotection, and DN replacement.63 Transplantation of these cells into the nigrostriatal pathway of parkinsonian rats and non-human primates treated with MPTP is associated with histological and functional recovery, inducing dopaminergic fibre sprouting in the pathway, with no procedure-related adverse events.62,64,65
Induced pluripotent stem cells are able to generate DNs specific to each patient, without ethical or immunological problems, and can be obtained from a wide range of sources; however, they do present issues related to mutagenicity, damage to genome integrity, and teratoma formation.45,66,67
Induced pluripotent stem cell modelsHuman stem cells are undifferentiated cells with the ability to self-renew and differentiate to different cell lines, derived from the 3 germ layers: the endoderm, mesoderm, and ectoderm.68 Some authors have classified stem cells into 2 groups: embryonic cells, and adult or somatic cells. Each type presents different levels of potentiality, and may be pluripotent, multipotent, and/or tissue progenitor cells.69 Embryonic pluripotent stem cells are derived from the embryoblast, the inner mass of the blastocyst, and are able to differentiate into any type of cell present in adults, with the exception of extraembryonic tissues.70
The new technology in stem cell therapies is iPSCs; these cells are derived from such somatic cells as fibroblasts, which are reprogrammed to a pluripotent state using Yamanaka factors (Oct3/4, Sox2, c-Myc, and Klf4).71 From this new state, iPSCs can differentiate to any type of cell present in adults.72 These cells have great potential for in vitro modelling of such neurodegenerative diseases as PD.73
Generation of dopaminergic neuronsDNs can be generated from adult cells reprogrammed in vitro to a pluripotent state. It is also possible to genetically modify iPSCs to study the effect of a specific gene using established neural differentiation protocols that in the majority of cases use morphogens and factors expressed in the normal development of DNs (Sonic Hedgehog, fibroblast growth factor 8, brain-derived neurotrophic factor, etc). The cells obtained can be used in vitro to create cell models mimicking the pathophysiology of PD, which may be useful in the development of neuroprotective molecules for future treatments.74–76 One of the major advantages of these models is their compatibility with the genetic modification techniques currently available: zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated endonuclease (CRISPR/Cas9) (Fig. 1).77
 models of Parkinson’s disease (PD). PD models using iPSCs begin with the acquisition of somatic cells from patients with the disease. After cultures of somatic cells (eg, fibroblasts) are established, cells are reprogrammed using different vectors (eg, Sendai virus, lentivirus, retrovirus, and the transcription factors Oct3/4, Sox2, c-Myc, or Klf-4) to obtain iPSCs. After successful reprogramming, cells are differentiated to neuronal lineage through the addition of transcription factors (eg, Sonic Hedgehog, fibroblast growth factor 8, or brain-derived neurotrophic factor). During the pluripotent state, iPSCs may also be genetically modified to overexpress or inhibit genes of clinical interest. Finally, the dopaminergic neurons generated are used to model PD with in vivo or in vivo techniques or cryopreserved for future use, targeting the development of novel treatments.")
Diagram summarising the generation of induced pluripotent stem cell (iPSC) models of Parkinson’s disease (PD).
PD models using iPSCs begin with the acquisition of somatic cells from patients with the disease. After cultures of somatic cells (eg, fibroblasts) are established, cells are reprogrammed using different vectors (eg, Sendai virus, lentivirus, retrovirus, and the transcription factors Oct3/4, Sox2, c-Myc, or Klf-4) to obtain iPSCs. After successful reprogramming, cells are differentiated to neuronal lineage through the addition of transcription factors (eg, Sonic Hedgehog, fibroblast growth factor 8, or brain-derived neurotrophic factor). During the pluripotent state, iPSCs may also be genetically modified to overexpress or inhibit genes of clinical interest. Finally, the dopaminergic neurons generated are used to model PD with in vivo or in vivo techniques or cryopreserved for future use, targeting the development of novel treatments.
In the 20th century, the first experiments were conducted to understand and treat PD, taking different analytical approaches and contributing new information. Yamanaka’s discovery of iPSCs has led to an explosion in research on their applications in PD.72,78,79 The development of preclinical models using iPSCs has significantly increased over the last decade, with each model bringing us closer to understanding the pathophysiology of the disease.72,75 Research into the development of familial PD has focused on the study of genetic factors involved in the pathogenesis of the disorder (Table 2).79 Essentially, researchers aim to induce the characteristic motor symptoms of PD (bradykinesia, tremor, stiffness, and behaviour) and to establish the role of genes known to be involved in the disease, such as SNCA, LRKK2, PINK1, PARKIN, and GBA1.14,35,81–86
Genetic models of Parkinson’s disease using induced pluripotent stem cells. Effects of modification of genes involved in Parkinson’s disease pathogenesis in induced pluripotent stem cell models.
| Gene | Effect | Ref. |
|---|---|---|
| SNCA | Accumulation or overexpression of α-synuclein in DNs | Byers et al.84 |
| LRRK2 | Reduced neurite outgrowth | Shi et al.85 |
| Increased oxidative stress | ||
| Deterioration of iPSCs’ capacity for self-renewal and neuronal differentiation | ||
| PINK1/PARKIN | Abnormal mitophagy and autophagy phenotypes | Shaltouki et al.,86 Pickrell and Youle87 |
| Greater vulnerability to stress | ||
| GBA1 | High α-synuclein levels, autophagy, lysosomal defects, calcium homeostasis imbalance, reduced DA uptake and storage | Schöndorf et al.83 |
SNCA has been linked to the accumulation or overexpression of α-synuclein in DNs.84 Regarding LRRK2, loss of function of the protein has been shown to reduce neurite outgrowth, increase oxidative stress, and cause DNA damage; it also impairs the self-renewal capacity and neuronal differentiation of iPSCs.85 A correlation has been reported between the PINK1 and PARKIN genes, with both contributing to cellular and mitochondrial homeostasis in DNs. Furthermore, DNs derived from iPSCs with mutations in either of these genes present abnormal mitophagy and autophagy phenotypes, as well as increased vulnerability to stress.86,87 Finally, GBA1 mutations are closely linked to increased levels of α-synuclein, autophagy, lysosomal defects, calcium homeostasis imbalances, and decreased DA uptake and storage in iPSC-derived DNs. Furthermore, epigenetic alterations affect DNA methylation in patients, inducing errors in protein turnover and variations in cell morphology.88
Recently, iPSC cell lines have been derived from patients with hereditary or sporadic PD, offering the advantage that specific PD phenotypes are caused by patients’ genetic profiles from the earliest stages of the disease.74,89 The majority of cases of PD are sporadic, which makes establishing aetiology a challenge, as no specific genetic mutations related to this form of the disease have been identified80; therefore, this research has focused on the differentiation of iPSC-derived DNs from patients with sporadic PD.90
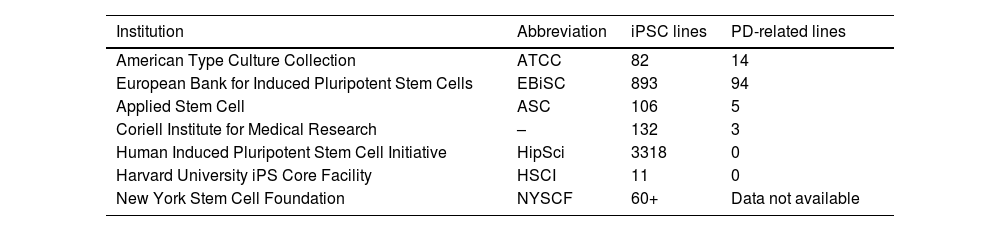
PD-related cell lines are found in numerous cell banks (Table 3). Various research projects have used these methods, with the creation of a library including over 60 iPSC lines developed through cell reprogramming with 3 different vectors: lentivirus, retrovirus, and Sendai virus.91,92
Data on cell banks storing cell lines, describing some of the best known institutions, the numbers of induced pluripotent stem cell lines available, and the number of cell lines related to Parkinson’s disease.
| Institution | Abbreviation | iPSC lines | PD-related lines |
|---|---|---|---|
| American Type Culture Collection | ATCC | 82 | 14 |
| European Bank for Induced Pluripotent Stem Cells | EBiSC | 893 | 94 |
| Applied Stem Cell | ASC | 106 | 5 |
| Coriell Institute for Medical Research | – | 132 | 3 |
| Human Induced Pluripotent Stem Cell Initiative | HipSci | 3318 | 0 |
| Harvard University iPS Core Facility | HSCI | 11 | 0 |
| New York Stem Cell Foundation | NYSCF | 60+ | Data not available |
Murine models using iPSCs have demonstrated the same potency and efficacy as DNs obtained from fetal tissue, showing high capacity for long-distance, target-specific axonal outgrowth and rapid, efficient, synchronised differentiation, avoiding tumour formation.93 It has recently been reported that these cells support functional recovery of lesion-induced deficits, positioning iPSCs as a promising future line of research in a broad range of diseases.8
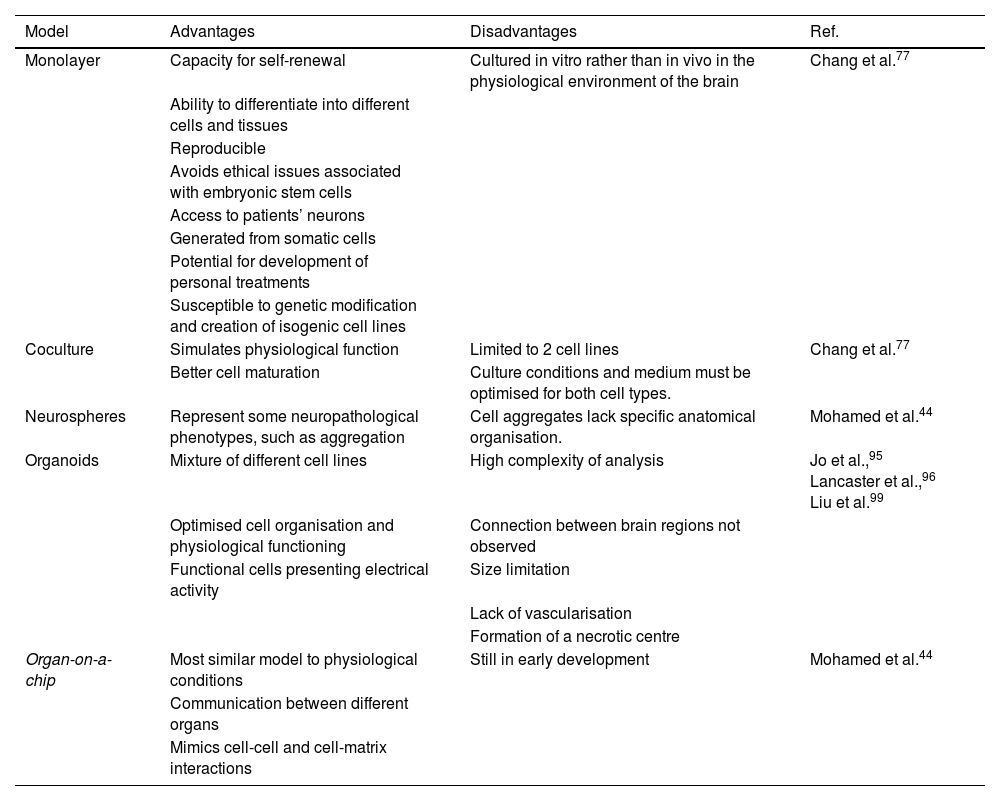
Monolayer cell growth models were the first to be developed, with a view to studying individual cellular and molecular mechanisms.52 However, such models may not be fully representative of the complexity of neurodegenerative diseases, leading to the development of coculture and 3-dimensional models (Table 4).77 Important advances have been made in coculture methods using 2 cell types (eg, astrocytes and neurons) to mimic cell activity in physiological conditions, with cell–cell interactions and a mixed extracellular matrix.52 The structural and metabolic support provided by astrocytes to neurons80 results in faster neuron maturation, higher levels of neural markers, and stabilisation of mitochondrial function due to reduced production of reactive oxygen species.94 The disadvantages of this method are the limitations on the cell lines that can be used and the need to optimise functional culture conditions for both types of cell.77
Models of Parkinson’s disease. Comparison of different induced pluripotent stem cell models of Parkinson’s disease and their respective advantages and disadvantages.
| Model | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Monolayer | Capacity for self-renewal | Cultured in vitro rather than in vivo in the physiological environment of the brain | Chang et al.77 |
| Ability to differentiate into different cells and tissues | |||
| Reproducible | |||
| Avoids ethical issues associated with embryonic stem cells | |||
| Access to patients’ neurons | |||
| Generated from somatic cells | |||
| Potential for development of personal treatments | |||
| Susceptible to genetic modification and creation of isogenic cell lines | |||
| Coculture | Simulates physiological function | Limited to 2 cell lines | Chang et al.77 |
| Better cell maturation | Culture conditions and medium must be optimised for both cell types. | ||
| Neurospheres | Represent some neuropathological phenotypes, such as aggregation | Cell aggregates lack specific anatomical organisation. | Mohamed et al.44 |
| Organoids | Mixture of different cell lines | High complexity of analysis | Jo et al.,95 Lancaster et al.,96 Liu et al.99 |
| Optimised cell organisation and physiological functioning | Connection between brain regions not observed | ||
| Functional cells presenting electrical activity | Size limitation | ||
| Lack of vascularisation | |||
| Formation of a necrotic centre | |||
| Organ-on-a-chip | Most similar model to physiological conditions | Still in early development | Mohamed et al.44 |
| Communication between different organs | |||
| Mimics cell-cell and cell-matrix interactions |
Source: Mohamed et al.44
A novel research method is the use of iPSCs to generate 3-dimensional organoids and neurospheres; with these techniques, iPSCs differentiate more spontaneously, over a period of 1–2 months, into functional neurons, more accurately modelling brain development and neurological disease.95,96 Organoids can be preserved in these cultures for up to a year, although they begin to shrink after 6 months.97 Limitations of these models include size restrictions, lack of vascularisation, short duration, the formation of a necrotic centre, imprecise identification of brain regions, variability between batches, and technical difficulty.98 These models are also used to study the physiological mechanisms involved in PD, to test potential treatments, and in the development of personalised medicine.99 Finally, organ-on-a-chip models use iPSCs in an in vitro microphysiological system in which several organoids are cocultured, with liquid flow providing contact between cells to mimic the physiological conditions of the body, including cell-cell and cell-matrix interactions; this promising method is currently in early development.44
Advantages of induced pluripotent stem cells over other modelsStem cells can be differentiated into SNpc DNs to model PD at the cellular level. However, iPSCs present the advantage that cells are patient-specific, avoiding the need for immunosuppression in transplantation and the ethical issues of acquiring human embryonic stem cells; furthermore, cells can be genetically corrected to produce functional phenotypes.100 The differentiation of iPSCs to DNs mimics the embryonic development of these cells, conserving the endogenous cellular machinery and transcription mechanisms.101 Additionally, it avoids the use of neurotoxins, maintaining natural development of the disease without stressful external stimuli.10
An area for further improvement in gene editing research is the optimal differentiation of DNs, which is dependent on molecular regulators of the function and stability of proteins involved in PD.102 The most widely used genetic modification techniques continue to present several limitations, including: 1) non-homologous end joining, which is prone to errors resulting in genomic instability and disease-causing mutations103; 2) homologous recombination using ZFN or TALEN presents targeting issues due to the limited availability of libraries for ZFN, and the size and complexity of vectors required for TALEN104; and 3) although there has been an explosion of progress in CRISPR/Cas9, its status as a novel technique means that analysis of Cas9 enzymes is required for correct targeting.105 Improving techniques for the editing of genes in specific tissues and cell populations in vivo continues to be one of the key challenges for safe, efficacious gene therapy.106
One of the difficulties of transplant-based cell therapy is the formation of Lewy bodies with the passage of time, leading to the reappearance of motor symptoms. This issue may be resolved by CRISPR/Cas9, a novel technology that has been used to develop iPSC-derived DNs with an SNCA deletion; in contact with pre-formed α-synuclein fibrils, these cells showed permanent resistance to the formation of α-synuclein aggregates.50
The first PD therapy based on the transplantation of iPSC-derived DN precursor cells into the SNpc began in 2018. The authors selected 7 patients with moderate PD; the first patient has presented no adverse reactions and, if this continues to be the case, the trial is expected to continue with the remaining patients this year.107,108 Before clinical studies were started in human patients, tests were conducted in mice to analyse tumorigenicity, toxicity, biodistribution of the DNs obtained, and teratoma formation; in a rat model with 6-OHDA lesions, animals showed a reduction of rotational asymmetry to normal levels; finally, survival of engrafted neurons was demonstrated in non-human primates, with no adverse effects.109 This project represents a great advance in the development of treatments for PD, whose success would lay new paths for treatment of the disease in the future.
Currently, 2 ongoing clinical studies registered with the United States National Institutes of Health are researching applications of iPSCs in patients with PD. The National Institutes of Health Clinical Center (National Heart, Lung, and Blood Institute) began a clinical study in June 2010 named “Characterization of patients with uncommon presentations and/or uncommon diseases associated with the cardiovascular system” (clinicaltrials.gov identifier: NCT01143454). The study aims to characterise the molecular aetiology, pathophysiology, and history of known and unknown rare diseases, including PD, which present with signs and symptoms associated with the risk of potential or manifest cardiovascular dysfunction, using biological materials and tissue samples to perfect diagnostic protocols.110
The second study, entitled “Development of iPS from donated somatic cells of patients with neurological diseases” (clinicaltrials.gov identifier: NCT00874783) has been in development since April 2009 by the Hadassah Medical Organisation, and aims to develop human iPSCs from cell cultures from patient skin biopsy or hair samples, using forced expression of transcription factors. The resulting cells will mainly be used to model such neurodegenerative diseases as PD for drug testing, to generate valuable information for basic research, and to develop technologies that may eventually enable the use of iPSCs in future transplantation therapies.111
Preclinical models based on iPSCs present certain limitations, such as the need for improved standardisation of protocols for iPSC acquisition, reprogramming, and differentiation; safety and administration to patients; the potential for tumour and teratoma formation; and the need to develop faster, more efficient, non-integrating induction methods.112 Despite these limitations, iPSC models currently present the greatest phenotypic similarity to PD, enabling researchers to study the cellular effects of mutations in real time, quantify the cellular and mitochondrial effects of oxidative stress, and analyse drugs and molecules with neuroprotective potential.10,113–116
Discussion and future perspectivesPD is a globally relevant disorder whose aetiology is not fully understood as the sporadic form is primarily multifactorial, and for which no effective treatments are available. Despite the fact that the available medications increase DA production in SNpc DNs, they only attenuate the symptoms of the disease, and do not reduce or prevent its progression.
Induced pluripotent stem cells may provide a fundamental model for research into such neurodegenerative diseases as PD, enabling testing of potential future treatments. DNs derived from these cells are able to reproduce the development of PD, and may therefore be used to study the progression of the disease and to identify molecular markers of potential diagnostic value. A wide range of options are available for developing models of PD, either with toxins or in vivo, in vitro, or coculture genetic techniques, and several animal species can be used.
It should be stressed that the standards required for the use of iPSCs in grafts to treat PD are yet to be perfected, in both the clinical and research spheres. One of the major challenges for iPSC models is the fact that whereas PD mainly presents in elderly individuals, reprogramming of somatic cells generates an embryonic-like state; in vitro models should therefore seek to reproduce the characteristics of neuronal senescence.
However, with tools for epigenetic modification, chromatin remodelling, and genomic regulation of genes relevant to PD, iPSC models are paving the way to personalised medicine. Patient cell lines could be generated from somatic cells, bypassing the ethical issues associated with the use of embryonic stem cells and enabling specific in vitro testing of drugs for individual patients.
FundingThis study was supported by the Mexican National Council of Science and Technology (CONACYT: project codes 300638 and 271307; FODECIJAL: project code 8084-2019).
Conflicts of interestNone.
