



Parkinson's disease (PD) is the second most common neurodegenerative disorder. It is characterised by selective loss of dopaminergic neurons in the substantia nigra pars compacta, which results in dopamine depletion, leading to a number of motor and non-motor symptoms.
DevelopmentIn recent years, the development of new animal models using nuclease-based genome-editing technology (ZFN, TALEN, and CRISPR/Cas9 nucleases) has enabled the introduction of custom-made modifications into the genome to replicate key features of PD, leading to significant advances in our understanding of the pathophysiology of the disease.
ConclusionsWe review the most recent studies on this new generation of in vitro and in vivo PD models, which replicate the most relevant symptoms of the disease and enable better understanding of the aetiology and mechanisms of PD. This may be helpful in the future development of effective treatments to halt or slow disease progression.
La enfermedad de Parkinson (EP) es el segundo trastorno neurodegenerativo más común, caracterizado por la pérdida selectiva de neuronas dopaminérgicas en la substancia nigra pars compacta, produciendo depleción en los niveles de dopamina y dando como resultado las manifestaciones clínicas de la enfermedad que se pueden clasificar como síntomas motrices y no motrices.
DesarrolloEn los últimos años, la generación de nuevos modelos animales basados en los sistemas de edición genética por nucleasas: ZFN, TALEN, CRISPR/Cas9, permiten la realización de modificaciones personalizadas en el genoma, replicando características clave que definen a la EP y consecuentemente avances significativos en la comprensión del proceso fisiopatológico de este trastorno.
ConclusiónEn esta revisión recopilamos los estudios más novedosos de esta nueva generación de modelos in vitro e in vivo de la EP que permiten emular síntomas clave y tener una mayor comprensión de la etiología o los mecanismos involucrados en el proceso de iniciación o desarrollo de la enfermedad y la futura prueba de terapias realmente eficaces para detener o ralentizar su progresión.
Parkinson's disease (PD) is the second most frequent neurodegenerative disease after Alzheimer disease, affecting 1% of individuals aged over 55 years and up to 4% of those aged over 80.1,2 Incidence is estimated at 8-18 cases per 100000 person-years.3 This progressive neurodegenerative condition causes excessive selective loss of dopaminergic neurons (DN) (50%-70%) in the substantia nigra pars compacta (SNPC), leading to a significant decrease in levels of dopamine reaching the striatum and consequently in functional impairment of the motor circuit.1,4 PD is characterised by a range of physiological symptoms, including resting tremor, muscle rigidity, bradykinesia, gait alterations, and postural instability.5 However, these motor manifestations can also be accompanied by such non-motor symptoms as olfactory deficits (hyposmia), sleep disorders, cognitive impairment, depression, fatigue, and pain.6,7
While the pathophysiological process of PD is not fully understood, a range of animal models of striatal damage have shed light on the aetiological and pathological factors and the molecular mechanisms involved; these models use rodent and primate species,4 as well as non-mammal models including the zebrafish,8 the nematode Caenorhabditis elegans,9 and the fruitfly Drosophila melanogaster.10 Neurodegeneration may be related to several different mechanisms, including oxidative stress, excessive free radical generation, environmental factors, genetics, and endogenous neurotoxins.11,12 Recent studies have also identified mutations in numerous genes in familial PD, which may play a role in the onset or development of the disease; these genes include the α-synuclein gene (SNCA), Parkin (PRKN/PARK2), PINK1, LRRK2, PARK7, DJ-1, GBA, UCH-L1, and MAPT/STH; such mutations may also be related to sporadic PD.11,13,14
Since the 1980s, mouse strains have been used as models for the study of the genetic, environmental, and pharmacological aspects of human diseases.15,16 Modern technology has greatly increased the speed and efficiency with which mutant strains can be generated.17 In the field of molecular biology, the last 20 years have seen the development of a number of technologies that can provoke double-strand breaks in DNA, enabling targeted genome editing. Three efficient, reliable gene editing methods currently exist: zinc finger nucleases (ZFN), transcription activator–like effector nucleases (TALEN), and most recently the CRISPR-Cas9 system (clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9).18 These techniques exploit natural DNA repair mechanisms, and can introduce modifications ranging from single-nucleotide substitutions to deletions of large segments.
This review aims to gather the most internationally relevant studies on the use of endonucleases and the derived transgenic models of the pathological mechanisms of PD, and discusses their relevance to the understanding of this neurodegenerative disease.
New techniques for designing transgenic modelsGiven the well-established fact that PD can be of either sporadic (90%-95% of cases) or familial origin (5%-10%), researchers have sought to study each type independently using 2 different types of models: toxin-induced (sporadic) and genetic models (familial).19 In vitro models of PD have used several cell lines, aiming to identify therapeutic candidates for drug and toxicology trials. For instance, the SH-SY5Y neuroblastoma line and the PC12 pheochromocytoma lines replicate DN degeneration.20,21 Other studies use such immortalised cell lines as the Lund human mesencephalic (LUHMES), MN9D (mouse), and CSM14.1 lines (rat), and primary cultures from postnatal mesencephalic cells (mouse and rat).22 Despite the value of these models for mimicking DN degeneration, they do not present the typical genotype of familial PD. Previously, transgenic models of familial PD were generated using viral transfection (adeno-associated viruses), a fairly inefficient technique.23 In contrast, new genetic engineering technologies have enabled more efficient generation of transgenic models, with significant reductions in timeframes and expense.24
This review only addresses genetic models, given the great advances made in this field due to improvements in gene editing tools, which enable genes to be more clearly associated with specific functions. This has led to the development of more complete transgenic models of PD.
Zinc finger nucleasesZFNs are enzymes that are able to cut double-stranded DNA at a specific site.
It is now possible to design zinc finger domains to target specific DNA sequences, enabling ZFNs to locate unique sequences within complex genomes.25
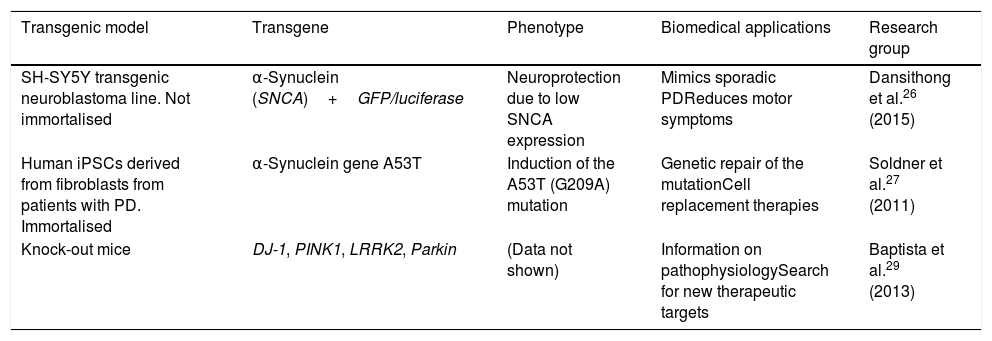
This technology has been used to create transgenic cellular models of PD, such as cell lines that express α-synuclein fused to the reporter genes luciferase or GFP, with the objective of testing molecules that may limit α-synuclein expression.26 The technique has also been used to perform genetic repair of the A53T mutation of SNCA in human induced pluripotent stem cells (iPSC) derived from patients with PD.27 Likewise, Sanders et al.28 used ZFN genome editing on the G2019S mutation of LRRK2 in human fibroblast-derived iPSCs, detecting no mitochondrial damage in differentiated progenitor and neural cells. The Michael J. Fox Foundation and the Sanger Institute have used the technique to produce several transgenic models of PD, creating DJ-1, PINK1, LRRK2, and Parkin knock-out rats, which display distinctive phenotypes of PD, providing pathophysiological information and contributing to the search for new therapeutic targets and drugs.29Table 1 summarises several transgenic models generated using ZFNs.
Transgenic cell and animal models of Parkinson's disease generated using zinc finder nuclease editing.
| Transgenic model | Transgene | Phenotype | Biomedical applications | Research group |
|---|---|---|---|---|
| SH-SY5Y transgenic neuroblastoma line. Not immortalised | α-Synuclein (SNCA)+GFP/luciferase | Neuroprotection due to low SNCA expression | Mimics sporadic PDReduces motor symptoms | Dansithong et al.26 (2015) |
| Human iPSCs derived from fibroblasts from patients with PD. Immortalised | α-Synuclein gene A53T | Induction of the A53T (G209A) mutation | Genetic repair of the mutationCell replacement therapies | Soldner et al.27 (2011) |
| Knock-out mice | DJ-1, PINK1, LRRK2, Parkin | (Data not shown) | Information on pathophysiologySearch for new therapeutic targets | Baptista et al.29 (2013) |
iPSC: induced pluripotent stem cell; PD: Parkinson's disease.
Specific programmable nucleases have a broad range of applications, using the dimeric FokI nuclease to induce personalised modifications of target sequences in the genome.
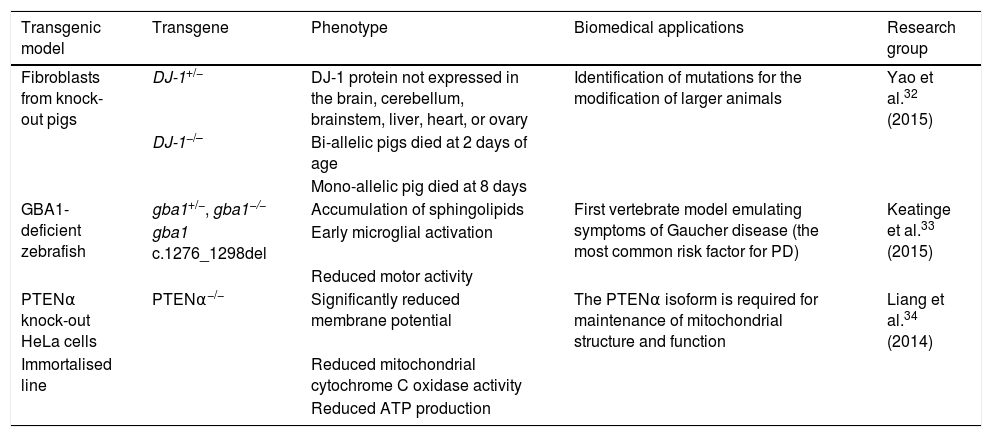
In particular, TALEN is a powerful genome editing tool that can cleave unique genetic sequences in live cells and in such organisms as zebrafish, rats, and pigs.30–32 Used in synergy with the somatic cell nuclear transfer technique, this editing tool has enabled the creation of bi-allelic DJ-1 knock-out pigs. Yao et al.32 obtained one DJ-1+/− and 2 DJ-1−/− piglets, in which sequencing and Western blot analysis showed that the DJ-1 protein was not expressed in any of the tissues studied (brain, cerebellum, brainstem, liver, heart, and ovary), indicating that the model was generated successfully. Mutations of the glucocerebrosidase 1 (GBA1) enzyme constitute another risk factor involved in PD development. Keatinge et al.33 used the TALEN system to generate a heterozygous zebrafish model of GBA1 deficiency (gba1+/−), which at 5 days post-fertilisation displayed marked sphingolipid accumulation and early microglial activation, as well as pronounced DN degeneration from week 8 and a considerable reduction in motor activity at 12 weeks. The PTEN protein has been shown to be critical in maintaining mitochondrial homeostasis, which is associated with proper neuronal function. Liang et al.34 recently identified the PTENα isoform of the protein. The researchers used the TALEN system to generate PTENα−/− knock-out HeLa cells, reporting significant reductions in membrane potentials, mitochondrial cytochrome C oxidase activity, and ATP production; this demonstrates that the PTENα isoform is necessary in maintaining mitochondrial structure and function. Table 2 summarises several transgenic models generated using TALEN.
Transgenic cell and animal models of Parkinson's disease generated using transcription activator-like effector nucleases.
| Transgenic model | Transgene | Phenotype | Biomedical applications | Research group |
|---|---|---|---|---|
| Fibroblasts from knock-out pigs | DJ-1+/− | DJ-1 protein not expressed in the brain, cerebellum, brainstem, liver, heart, or ovary | Identification of mutations for the modification of larger animals | Yao et al.32 (2015) |
| DJ-1−/– | Bi-allelic pigs died at 2 days of age | |||
| Mono-allelic pig died at 8 days | ||||
| GBA1-deficient zebrafish | gba1+/−, gba1−/− | Accumulation of sphingolipids | First vertebrate model emulating symptoms of Gaucher disease (the most common risk factor for PD) | Keatinge et al.33 (2015) |
| gba1 c.1276_1298del | Early microglial activation | |||
| Reduced motor activity | ||||
| PTENα knock-out HeLa cells | PTENα−/− | Significantly reduced membrane potential | The PTENα isoform is required for maintenance of mitochondrial structure and function | Liang et al.34 (2014) |
| Immortalised line | Reduced mitochondrial cytochrome C oxidase activity | |||
| Reduced ATP production |
ATP: adenosine triphosphate; PD: Parkinson's disease.
iPSCs are generated by reprogramming differentiated somatic cells to pluripotency through the ectopic coexpression of transcription factors (Oct4, Sox2, Klf4, and c-Myc).35 The reprogrammed cells are capable of self-renewal and differentiation into different cell lines, including neurons.36 This makes them an ideal model for in vitro simulation of numerous pathological processes, including such neurodegenerative diseases as PD, with a view to the development of novel, effective treatments, such as cell replacement therapy.37
It is now possible to obtain iPSCs from numerous organs and tissues, including cells from the stomach and liver, neural stem cells, peripheral blood cells, and hair follicle keratinocytes; in the study of neurodegenerative diseases in particular, fibroblasts are obtained from biopsies of patients for subsequent reprogramming into neurons and correction with nuclease enzymes to assess their functionality.38,39
Another gene editing method is the revolutionary CRISPR-Cas9 system, in which RNAs guide the Cas9 enzyme to the target DNA.40 The system originally belonged to a prokaryotic adaptive immune mechanism, first described in Escherichia coli.41 The CRISPR system is found in a non-coding form, which is subsequently transcribed into short RNA sequences (CRISPR RNA [crRNA]) separated by spacer sequences of similar size (20-50bp).42 These RNAs direct the Cas9 nuclease to specific sequences of genomic DNA, upstream from a protospacer adjacent motif (PAM) sequence (NGG), which occurs on average every 8bp in the human genome.43 Subsequently, the Cas9 nuclease precisely cuts both strands of DNA and the damage is repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR), interrupting and inactivating the target gene. In 2012, Jinek et al.44 fused a crRNA containing the guide sequence to a trans-activated crRNA (tracrRNA), generating a simple guide RNA (sgRNA), which facilitates DNA cleavage by CRISPR-Cas9. The system has also been simplified to use a single chimeric sgRNA; in this simplified system, Cas9 is guided by an sgRNA (crRNA-tracrRNA) of 20 nucleotides in length, the maximum length for specific recognition of the target gene. CRISPR-Cas9 has been used in the genetic engineering of practically all types of cells and organisms, enabling the generation of organisms with multiple mutations or large chromosomal deletions.45
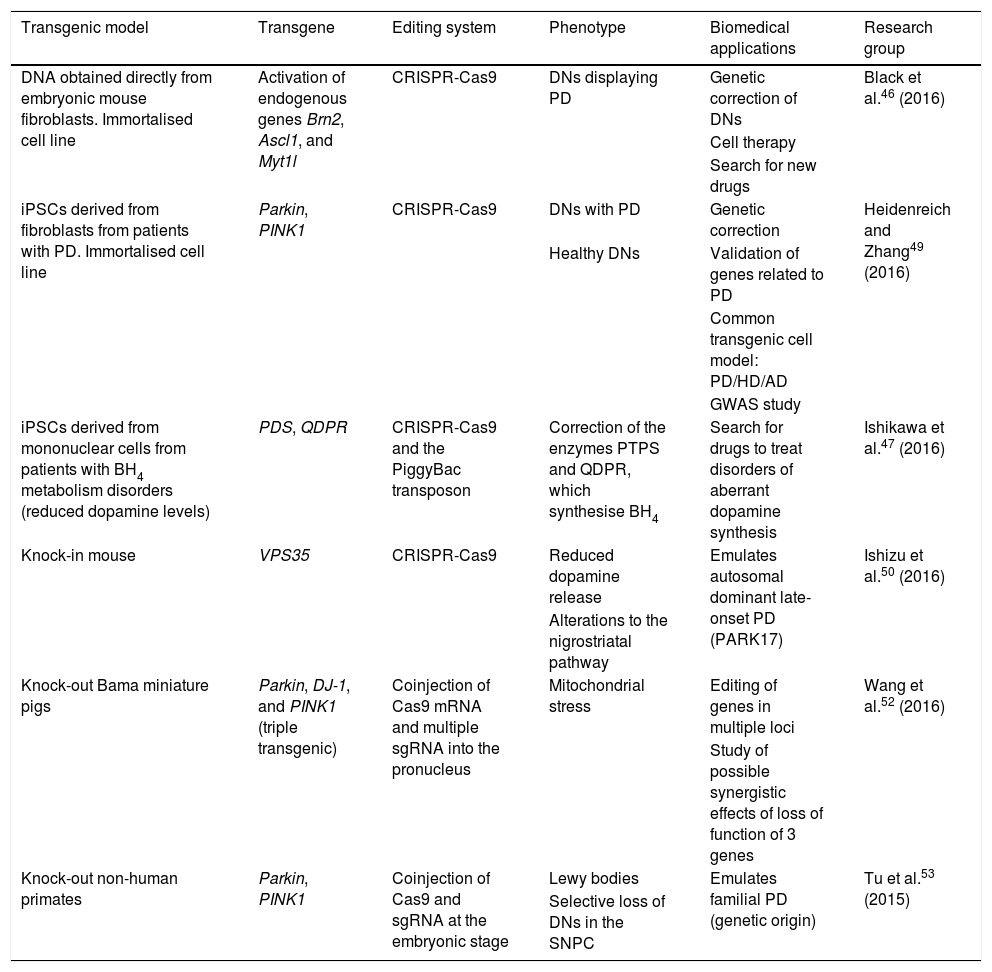
Combination of technologies: induced pluripotent stem cells and the CRISPR-Cas9 systemIn line with the above, various research groups have begun using the CRISPR-Cas9 system to obtain iPSCs for modelling PD. One novel application of CRISPR is in improving the efficiency of the direct reprogramming of fibroblasts into neurons. Black et al.46 recently reported using the CRISPR-Cas9 system to convert mouse embryonic fibroblasts directly into neurons by epigenetic activation of endogenous genes (Brn2, Ascl1, and Myt1l) at the target locus, inducing endogenous expression which persists during reprogramming. Epigenetic activation and remodelling of native chromatin promotes cellular reprogramming without the forced overexpression caused by the integration of plasmids into the genome, as in traditional methods of ectopic overexpression; the system therefore represents a new alternative, surpassing epigenetic barriers in cell fate determination. In other studies, CRISPR has been used for genome-level correction of iPSCs. Significant decreases in dopamine levels have been observed in the context of tetrahydrobiopterin (BH4) deficiency; this process has been linked to PD. Ishikawa et al.47 used CRISPR-Cas9 and the PiggyBac transposon system to genetically correct the genes encoding enzymes for BH4 synthesis in iPSCs derived from mononuclear peripheral blood cells from patients with 6-pyruvoyltetrahydropterin synthase and dihydropteridine reductase deficiencies. Correction of the genes after differentiation of iPSCs to DNs restored extracellular dopamine production and BH4 and tyrosine hydroxylase levels.
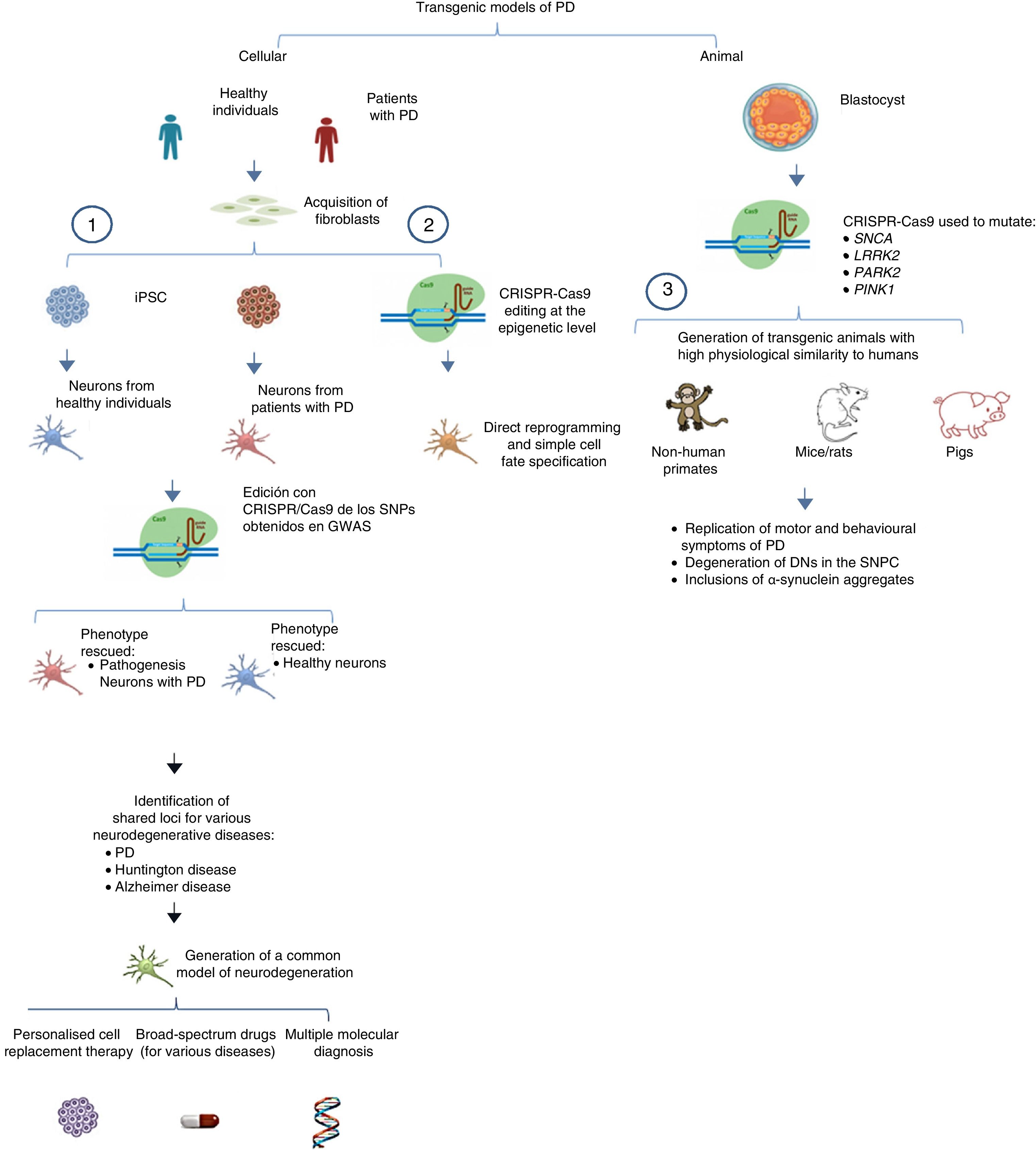
Another important application of CRISPR-Cas9 in iPSCs is in genome-wide association studies (GWAS) that evaluate single-nucleotide polymorphisms and attempt to identify causes of disease (in this case, PD) through population-scale sequencing.48 Heidenreich and Zhang49 conducted a three-part GWAS-based study using iPSCs derived from fibroblasts from healthy individuals and patients with PD, differentiating the cells into neurons in vitro. (1) Patient-derived iPSCs are edited with CRISPR-Cas9, targeting the specific site of homologous recombination of genes thought to be involved in pathogenesis (candidate genes). If disease phenotype is rescued and the phenotype after editing is similar to that of neurons derived from healthy individuals, the candidate gene is confirmed to be valid. (2) Suspected candidate genes in neurons derived from healthy individuals are mutated using CRISPR-Cas9; if pathogenesis of PD is recapitulated in vitro, the candidate gene is confirmed. (3) This supports the association between genes with multiple shared loci and such diseases as PD and Alzheimer disease. The CRISPR system can be used to identify correlations between functional phenotypes and different genetic mutations through the application of sgRNAs. This information may soon lead to the generation of transgenic cellular models with shared loci for various neurodegenerative diseases (e.g., Alzheimer disease, PD, Huntington disease), potentially leading to studies searching for molecules associated with pathogenesis or multiple functional therapies for these diseases. Table 3 summarises several transgenic models generated using CRISPR-Cas9.
Transgenic cell and animal models of Parkinson's disease generated using the CRISPR-Cas9 system.
| Transgenic model | Transgene | Editing system | Phenotype | Biomedical applications | Research group |
|---|---|---|---|---|---|
| DNA obtained directly from embryonic mouse fibroblasts. Immortalised cell line | Activation of endogenous genes Brn2, Ascl1, and Myt1l | CRISPR-Cas9 | DNs displaying PD | Genetic correction of DNs | Black et al.46 (2016) |
| Cell therapy | |||||
| Search for new drugs | |||||
| iPSCs derived from fibroblasts from patients with PD. Immortalised cell line | Parkin, PINK1 | CRISPR-Cas9 | DNs with PD | Genetic correction | Heidenreich and Zhang49 (2016) |
| Healthy DNs | Validation of genes related to PD | ||||
| Common transgenic cell model: PD/HD/AD | |||||
| GWAS study | |||||
| iPSCs derived from mononuclear cells from patients with BH4 metabolism disorders (reduced dopamine levels) | PDS, QDPR | CRISPR-Cas9 and the PiggyBac transposon | Correction of the enzymes PTPS and QDPR, which synthesise BH4 | Search for drugs to treat disorders of aberrant dopamine synthesis | Ishikawa et al.47 (2016) |
| Knock-in mouse | VPS35 | CRISPR-Cas9 | Reduced dopamine release | Emulates autosomal dominant late-onset PD (PARK17) | Ishizu et al.50 (2016) |
| Alterations to the nigrostriatal pathway | |||||
| Knock-out Bama miniature pigs | Parkin, DJ-1, and PINK1 (triple transgenic) | Coinjection of Cas9 mRNA and multiple sgRNA into the pronucleus | Mitochondrial stress | Editing of genes in multiple loci | Wang et al.52 (2016) |
| Study of possible synergistic effects of loss of function of 3 genes | |||||
| Knock-out non-human primates | Parkin, PINK1 | Coinjection of Cas9 and sgRNA at the embryonic stage | Lewy bodies | Emulates familial PD (genetic origin) | Tu et al.53 (2015) |
| Selective loss of DNs in the SNPC |
AD: Alzheimer disease; BH4: tetrahydrobiopterin; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9; DN: dopaminergic neuron; GWAS: genome-wide association study; HD: Huntington disease; iPSC: induced pluripotent stem cell; PD: Parkinson's disease; sgRNA: single guide RNA; SNPC: substantia nigra pars compacta.
The CRISPR system has been put to effective use in the creation of animal models of PD, with studies aiming to shed light on the natural process of the disease in association with point mutations and the development of new treatments. The CRISPR-Cas9 system is used today to generate knock-in models, such as the Vps35 D620N transgenic mouse (used to study the pathogenesis of the PARK17 adult-onset form of the disease, associated with point mutations in the vacuolar protein sorting 35 gene); these mice express the homologous mutant protein, and neither heterozygous nor homozygous mice die prematurely or present clear neurodegeneration before 70 weeks of age.50 Another study evaluates the PARK2 and PINK1 genes, involved in autosomal recessive PD, using CRISPR-Cas9 and the somatic cell nuclear transfer technique in a domestic pig model; the researchers report an approximate success rate of 38% in obtaining PARK2−/−/PINK1−/− double-knock-out homozygous cell colonies.51 Viable transgenic piglets did not present typical symptoms (agitation, rigidity, slow movement, difficulty walking, etc.) in a 7-month observation period.51 Wang et al.52 recently obtained a (knock-out) model of human PD in Bama miniature pig pronuclear embryos through in vivo injection of Cas9 mRNA and multiple sgRNAs, targeting 3 genes: Parkin, DJ-1, and PINK1. The researchers demonstrated a robust capacity to edit multiple genes in a transgenic model, with 100% efficiency, without inducing mutations in significant, non-specific sites (off-target events). Pigs did not show typical symptoms of PD at 10 months of age, which the authors attribute to a synergistic effect of the inactivation of all 3 genes. The simplicity of modifying multiple genes in pigs using CRISPR-Cas9 is of great future value to medicine and agriculture.
The system can also be used to edit these genes in non-human primates and other large animals, inactivating genetic expression. When both alleles are mutated (null−/−), Parkin and PINK1 lose all functionality, resulting in a model of familiar PD. To ensure that expression of these alleles is disrupted, multiple target regions can be designed, with fertilised eggs being injected with several sgRNAs and Cas9 at the one-cell stage.53 Following this procedure, transgenic non-human primates can be made to display such complex motor symptoms as bradykinesia, tremor, rigidity, and postural instability, as well as Lewy bodies, which are characteristic of patients with PD (Fig. 1).54Table 3 summarises some models generated using CRISPR-Cas9.
 Selection of candidate genes for Parkinson")
Transgenic models of PD generated with CRISPR-Cas9.
(1) Selection of candidate genes for Parkinson's disease using CRISPR-Cas9 in neurons differentiated from fibroblast-derived induced pluripotent stem cells from patients in genome-wide association studies, and future applications44 (adapted from Heidenrich and Zhang49).
(2) Direct reprogramming of fibroblasts to dopaminergic neurons through epigenetic manipulation with CRISPR-Cas9. The lack of ectopic overexpression simplifies cell fate specification.
(3) Generation of transgenic animal models by gene editing in embryos, associated with familial PD. These models can reproduce the pathophysiological and behavioural characteristics of PD.
CRISPR-Cas9: clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9; DN: dopaminergic neuron; iPSC: induced pluripotent stem cell; PD: Parkinson's disease; SNPC: substantia nigra pars compacta.
As mentioned above, one of the most relevant characteristics of CRISPR-Cas9 is its capacity to generate mutations in almost any region of the genome.55–57 This is a considerable advantage in the generation of transgenic models of PD in larger animals, as co-injection of Cas9 and sgRNA greatly simplifies induction of the mutations of interest, which has represented a great challenge in the past. Horii et al.58 evaluate 3 microinjection techniques for the generation of transgenic models in mouse embryos: injecting DNA into the pronucleus, injecting RNA into the pronucleus, and injecting RNA into the cytoplasm. They conclude that the latter technique is the most effective at the blastocyst stage and for producing pups, and show that the other 2 techniques are detrimental to embryo viability. This is an important factor to consider in the generation of functional transgenic mouse models through the use of CRISPR-Cas9 at the embryo stage.
In addition to the transgenic models generated using nucleases, other recent models specifically focus on altering transcription factors essential to the development of mature DNs, promoting cell loss in the substantia nigra pars compacta. The most relevant models are discussed below.
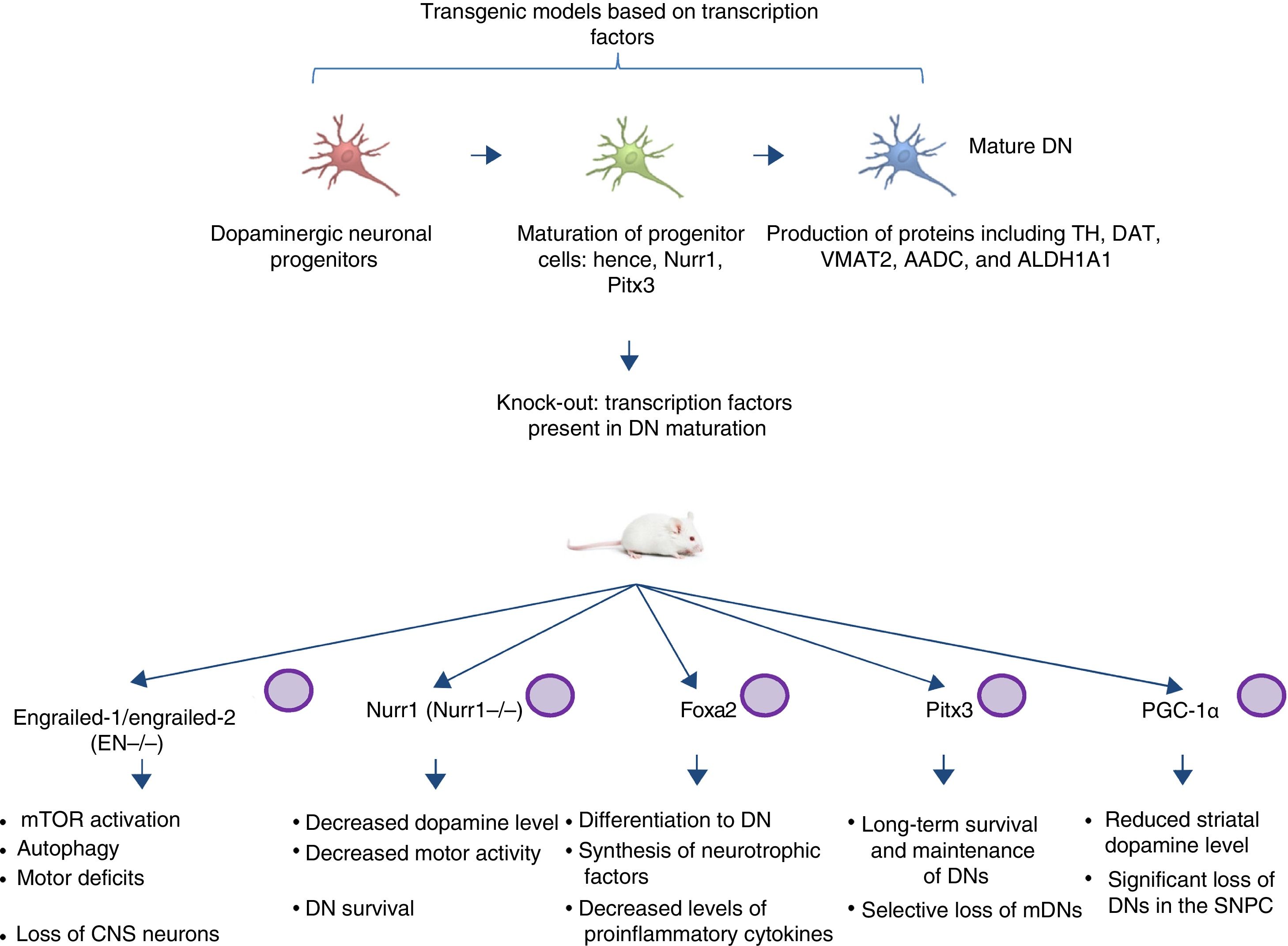
Transgenic models of Parkinson's disease derived from transcription factors necessary to dopaminergic neuron developmentAnother emerging sector in PD modelling is the development of mice with mutations targeting transcription factors important to the generation, development, and maintenance of mature DNs (Fig. 2). Various studies have clarified the genetics and signalling pathways of the processes controlling mature DN generation,59 with the expression of engrailed-1/engrailed-2 (EN−/−), Foxa1/2, Nurr1, Otx2, and Pitx3 (among others) being involved in lifelong maintenance of midbrain DNs (mDN).60,61 The most important examples include the association between these transcription factors and neuroprotective functions in PD; for instance, Otx2 expression prevents mDN loss in heterozygous EN1+/− mice.62 The engrailed homeoprotein is involved in activation of the mTOR pathway (an essential pathway in the cell) and in autophagy regulation.63 It also protects mDNs against MPTP through translation of subunits of mitochondrial complex I, improving their activity and increasing ATP synthesis.64,65 This demonstrates the importance of the translation of mRNA that correspond to mitochondrial components; many studies have shown that failure to meet the high energy requirements of mDNs is directly related to a considerable part of PD pathogenesis.66,67 Another knock-out mouse model involves modification of Nurr1, which is regulated by mutant α-synuclein68; this is thought to compromise glial cell line-derived neurotrophic factor (GDNF)/RET signalling, which contributes to the survival of DNs and other neuron types and has been proposed as a potential therapeutic target in PD.69 A recent study demonstrated that forced expression of Nurr1 and Foxa2 (which is essential in DN specification and differentiation) in glial cells protects mDNs in a mouse MPTP model of PD.70,71 These 2 genes display a synergistic mechanism of action in the microglia, with decreased levels of proinflammatory cytokines and increased synthesis of such neurotrophic factors as GDNF, BDNF, NT3, and SHH (among others), which regulate natural mechanisms of recovery, regeneration, and protection in nervous tissue.70
, Foxa1/2, Nurr1, Pitx3, and PGC-1α, the signalling pathways these factors participate in, and the pathophysiology (phenotype) caused by knock-out of these genes. CNS: central nervous system; DN: dopaminergic neuron; mDN: midbrain dopaminergic neuron; mTOR: mechanistic target of rapamycin; SNPC: substantia nigra pars compacta.")
Transgenic mouse models associated with transcription factors. These models employ mutant variants of transcription factors essential to the generation, development, and maintenance of mature dopaminergic neurons. The diagram lists the models generated through mutation of the transcription factors engrailed-1/engrailed-2 (EN−/−), Foxa1/2, Nurr1, Pitx3, and PGC-1α, the signalling pathways these factors participate in, and the pathophysiology (phenotype) caused by knock-out of these genes.
CNS: central nervous system; DN: dopaminergic neuron; mDN: midbrain dopaminergic neuron; mTOR: mechanistic target of rapamycin; SNPC: substantia nigra pars compacta.
Pitx3 is another factor that contributes considerably to mDN long-term survival and maintenance pathways. Filali and Lalonde72 generated a Pitx3-deficient mouse (the Aphakia mouse), observing decreased numbers of DNs in the substantia nigra and decreased tyrosine hydroxylase levels in the striatum. Pitx3 targets ALDH1A1, encoding the enzyme aldehyde dehydrogenase, which is essential to the production of retinoic acid and promotes antiapoptotic and antioxidant activity in mDN subpopulations73; this model therefore favours selective mDN loss, making it an ideal tool for PD research.
Various studies employing animal and in vitro models have found an association between peroxisome proliferator-activated receptor γcoactivator-1α (PGC-1α) receptor and PD. In an experiment with PGC-1α knock-out rats, Jiang et al.74 demonstrated significant DN loss and reduced dopamine levels in the striatum; these findings were directly related to the loss of PGC-1α and DNs in the SNPC. This clearly has significant implications for the generation of suitable transgenic models, as a direct relationship is established between the genetic knock-out and the phenotype associated with loss of function. Furthermore, it should be noted that the studies described above, using mouse models based on transcription factors, do not use the CRISPR-Cas9 system or other nucleases; these technologies may make an interesting contribution, facilitating the design of these transgenic models.
Models of Parkinson's disease employing nucleases related to ageingIt is well established that ageing plays a significant role in many neurodegenerative diseases, and is the main risk factor for developing PD.75,76 According to a thorough review by Reeve et al.,77 some changes related to age and ageing promote a hostile environment in the substantia nigra, leading to DN death. These processes include dopamine metabolism, accumulation of iron, calcium dynamics, mutations in mitochondrial DNA, neuromelanin accumulation, and reduced efficiency of protein degradation. Numerous studies report that these changes are sufficient to provoke neuronal loss, leading to PD in some cases. Studer et al.78 describe an experimental strategy for modelling ageing in in vitro models; these researchers discuss the induction of age-related parameters in populations of healthy and patient-specific cells without inducing toxicity. These paradigms may be a robust tool for measuring age-related changes, providing information on the interaction between genetic (isogenic lines) and environmental factors on disease phenotype. Another novel technique for generating models of ageing is the application of the new nuclease-based gene editing tools to directly manipulate key age-related molecular and cellular characteristics. CRISPR-Cas9 will greatly facilitate the targeting of signalling pathways or combinations of these pathways. A recent example was described by Harel et al.,79 who manipulated markers of ageing, including telomere shortening, epigenetic alterations, loss of proteostasis, and nutrient sensing (the TERT, ASH2L, ATG5, IGF1R, and RPS6KB1 genes) in the turquoise killifish, a short-lived African species, which represents a pipeline for new vertebrate models with a reduced timescale of ageing (6 months); such models show promise in the search for more reliable therapeutic targets related to signs of ageing. This represents a considerable advance, given the complexity of modelling ageing and neurodegeneration: a large majority of animal models develop the typical symptoms and die at a young age, whereas human patients with age-related disorders develop symptoms at an older age; this is a considerable obstacle to extrapolating findings to the clinical setting.80–82 Therefore, the study of ageing is an emerging area requiring the application of nucleases (ZFN, TALEN, and CRISPR-Cas9) in order to generate pipelines with high-throughput methods for identifying new therapeutic targets related to key characteristics involved in age-related neurodegenerative processes.
Disadvantages of the use of nucleases in generating transgenic modelsZinc finger nucleasesZFNs were part of the first generation of targeted gene editing tools, and have been used both in the generation of animal disease models and in treatments for human patients.83 However, they present significant disadvantages vis-à-vis other nucleases. The most important of these is the engineering of the tool itself: the assembly of zinc finger domains with a determined nucleotide sequence has proven to be a challenge.84 Another issue is the limited target site selection: ZFNs are able to recognise binding sites every 200bp in genomic DNA; this has been reduced to every 50bp in commercially available ZFNs.85 The generation of knock-out models is not affected by this, since ZFN can be used to introduce a frameshift in the early coding sequence, resulting in small, random insertions or deletions in any part of the gene and promoting loss of function. However, modifying specific sequences is relatively complex, making it an inefficient tool for replacing sequences in the genome (knock-in models).86–90
Transcription activator-like effector nucleasesTALEN was created 2 years before the CRISPR-Cas9 system, and artificial TALEN nucleases can theoretically be used to make double-strand DNA breaks anywhere in the genome with known recognition sites of DNA-binding domains.91 A considerable limitation of the technology is that it requires the presence of a thymine before the 5′ end of the target sequence. The binding site is selected through variations in the length of the spacer sequence. Interaction between the W232 residue at the N-terminal region of the DNA binding domain and the thymine N-terminal has been shown to affect TALEN's capacity to bind to the target site.92 However, these complications may be overcome through the production and selection of different mutant variants which have TALEN-N terminal domains capable of binding to adenine, cytosine, or guanine.
CRISPR systemDespite the many benefits of the CRISPR system, its editing efficiency is directly affected by several parameters inherent to the nuclease's mechanism of action. These include proper design of the sgRNAs, the method of delivering genetic material to the nucleus, the activity of the Cas9 enzyme, off-target effects, and the low incidence of the HDR mechanism in cells; issues have also been observed with the system's specificity, efficiency, and fidelity.93
Inadequate sgRNA design compromises the specificity and efficiency of targeted editing.94 A robust experimental design requires the generation of numerous sgRNAs, as several experiments have shown that some sgRNAs can be less efficient or even inactive.94–98 The order and composition of sgRNAs can have either a negative or a positive influence over the editing process. For instance, a guanine at the 5′ end of the sgRNA is required for expression of the U6 promoter.94 There should also be a guanine in the first or second position proximal to the PAM sequence, where the Cas9 enzyme selectively binds to the sequence; a cytosine in these positions is unfavourable to the process. Due to reduced expression of sgRNAs when multiple uracils are present, thymines are disfavoured in positions adjacent to the PAM.95 Guanine-rich sgRNAs containing low amounts of adenine display increased stability and efficiency.96 Off-target effects constitute another factor influencing specificity. Despite the improved specificity of the CRISPR-Cas9 system with respect to other nucleases, both in vitro and in vivo studies report multiple mutations in non-specific sites, which are extremely undesirable in biological studies, gene therapy, and targeted editing. Various studies report that Cas9-mediated double-strand DNA cleavage may be inhibited by a mismatch between the complementary region of the sgRNA and the target sequence, particularly in the nucleotide region adjacent to the PAM sequence.99,100 Whole-genome sequencing studies have found that mismatches in the terminal region of sgRNAs are better tolerated.99,100 Another study notes that in human cells, more than 5 mismatches can result in cleavage at off-target sites, which show similar frequencies of mutations to on-target sites.101
As mentioned previously, Cas9's action mechanism exploits natural cell repair pathways (NHEJ and HDR). Upon cleavage at the target site, various proteins are recruited for damage repair: KU70, DNA-PKcs, Artemis, XRCC4, pol μ, XLF/Cernunnos, KU80, and DNA ligase IV for NHEJ102; and RAD50, RAD51, MRE 11, Nbs1, XRCC2, XRCC3, RAD52, RAD54B, and BRCA2,102 as well as invasion by a homologous donor DNA template, for HDR.103–105 However, these mechanisms do not occur in equal proportions: NHEJ is more frequent than HDR; it is also more error-prone, and cannot be used to introduce nucleotide substitutions or for precise editing.93 Improving the incidence of HDR constitutes a challenge, but will increase the efficiency of CRISPR-Cas9, leading to improvements in the field of gene therapy and the generation of transgenic models.
ConclusionOne of the main challenges in PD is the shortage of models emulating pathophysiology due to DN neurodegeneration and proteinopathy, which results in unsuitable and ineffective treatments. Therefore, it is of great importance to generate a transgenic model accurately recreating the typical characteristics of the disease. Nuclease-based tools (ZFN, TALEN, and CRISPR-Cas9) offer the ability to generate models mimicking the characteristic symptoms, which may provide greater insight into the aetiology and molecular mechanisms involved in the onset and development of PD. This may contribute to novel studies into early diagnosis, new potential therapeutic targets, the selective correction of disease-causing genes or mutations, and the identification of molecules that may halt or slow disease progression. New technologies for reprogramming iPSCs offer promising options for overcoming barriers to cell fate specification and favouring the future possibility of personalised medicine, the effective search for novel drugs, and the application of cell replacement therapies in clinical practice, both for PD and for other complex diseases.
Conflicts of interestThe authors have no conflicts of interest to declare.
This project received funding from the CONACYT 2016 fund to support the strengthening and development of scientific and technological infrastructure (grant no. 271307) and the CONACyT Stem Cells and Regenerative Medicine Network.
Please cite this article as: Cota-Coronado JA, Sandoval-Ávila S, Gaytan-Dávila YP, Diaz NF, Vega-Ruiz B, Padilla-Camberos E, et al. Nuevos modelos transgénicos para el estudio de la enfermedad de Parkinson basados en sistemas de edición con nucleasas. Neurología. 2020;35:486–499.
