





La enfermedad de Parkinson (EP) es el segundo trastorno neurodegenerativo más común, caracterizado por la pérdida selectiva de neuronas dopaminérgicas en la substancia nigra pars compacta, produciendo depleción en los niveles de dopamina y dando como resultado las manifestaciones clínicas de la enfermedad que se pueden clasificar como síntomas motrices y no motrices.
DesarrolloEn los últimos años, la generación de nuevos modelos animales basados en los sistemas de edición genética por nucleasas: ZFN, TALEN, CRISPR/Cas9, permiten la realización de modificaciones personalizadas en el genoma, replicando características clave que definen a la EP y consecuentemente avances significativos en la comprensión del proceso fisiopatológico de este trastorno.
ConclusiónEn esta revisión recopilamos los estudios más novedosos de esta nueva generación de modelos in vitro e in vivo de la EP que permiten emular síntomas clave y tener una mayor comprensión de la etiología o los mecanismos involucrados en el proceso de iniciación o desarrollo de la enfermedad y la futura prueba de terapias realmente eficaces para detener o ralentizar su progresión.
Parkinson's disease (PD) is the second most common neurodegenerative disorder. It is characterised by selective loss of dopaminergic neurons in the substantia nigra pars compacta, which results in dopamine depletion, leading to a number of motor and non-motor symptoms.
DevelopmentIn recent years, the development of new animal models using nuclease-based genome-editing technology (ZFN, TALEN, and CRISPR/Cas9 nucleases) has enabled the introduction of custom-made modifications into the genome to replicate key features of PD, leading to significant advances in our understanding of the pathophysiology of the disease.
ConclusionsWe review the most recent studies on this new generation of in vitro and in vivo PD models, which replicate the most relevant symptoms of the disease and enable better understanding of the aetiology and mechanisms of PD. This may be helpful in the future development of effective treatments to halt or slow disease progression.
La enfermedad de Parkinson (EP) es la segunda enfermedad neurodegenerativa más común después de la enfermedad de Alzheimer. Afecta al 1% de la población mayor de 55años y hasta el 4% en la población mayor a 80años1,2, además presenta una incidencia de 8 a 18 personas al año por cada 100.000 habitantes3. Este padecimiento progresivo y neurodegenerativo resulta de una pérdida excesiva selectiva de neuronas dopaminérgicas (50-70%) de la substancia nigra pars compacta (SNPC), promoviendo una disminución significativa en los niveles de dopamina hacia el cuerpo estriado y, en consecuencia, el deterioro funcional del circuito motor1,4. La EP se caracteriza por varios síntomas fisiológicos, como temblor en reposo, rigidez muscular, bradicinesia, alteraciones al caminar e inestabilidad postural5. Sin embargo, estas manifestaciones motoras pueden ir acompañadas de síntomas no motores, como déficits olfativos (hiposmia), alteraciones del sueño, deterioro cognitivo, depresión, fatiga y dolor6,7.
Aunque el proceso fisiopatológico de la EP no está completamente dilucidado, se ha logrado una mejor comprensión sobre la etiología, la patología y los mecanismos moleculares involucrados gracias a diversos modelos animales que exhiben daño nigroestriatal en una variedad de especies tales como roedores y primates4, así como los modelos que no son mamíferos, incluyendo al pez cebra8, el nematodo Caenorhabditis elegans9 y la mosca de la fruta Drosophila melanogaster10. La neurodegeneración podría estar relacionada con diferentes mecanismos, entre los que destacan el estrés oxidativo, la excesiva formación de radicales libres, los factores ambientales, el factor genético y las neurotoxinas endógenas11,12. Por otra parte, estudios recientes han demostrado mutaciones en diversos genes de la EP familiar que probablemente participan en la iniciación o desarrollo de la enfermedad, tales como: α-sinucleína (SNCA), Parkina (PRKN/PARK2), PINK 1, LRRK2, PARK7, Dj-1, GBA, UCH-L1 y MAPT/STH, y que pudieran estar relacionados a su vez con la EP de tipo esporádico11,13,14.
Desde los años ochenta, las cepas de ratón se han utilizado como modelos para estudiar los componentes genéticos, medioambientales y farmacológicos de las enfermedades humanas15,16. Hoy en día existen nuevas tecnologías que están aumentando grandemente la eficiencia y la velocidad de generación de cepas mutantes17. Desde hace dos décadas, en el área de biología molecular se han desarrollado varias tecnologías que pueden generar rupturas sobre el ADN bicatenario, con la finalidad de hacer edición genómica dirigida. Actualmente existen tres métodos de edición génica que actúan de manera confiable y eficiente: las nucleasas con dedos de zinc (ZFN), las nucleasas efectoras tipo activador de la transcripción (TALEN) y recientemente la tecnología del sistema de Repeticiones Palindrómicas Cortas Agrupadas y Regularmente Interespaciadas en conjunto con la proteína asociada 9 (CRISPR/Cas9)18. Estas técnicas explotan los mecanismos naturales de reparación del ADN, y son capaces de generar desde la sustitución de un nucleótido hasta la eliminación de un segmento de gran tamaño.
Esta revisión tiene como objetivo recopilar los estudios más relevantes a nivel mundial sobre el empleo de endonucleasas y los modelos transgénicos derivados para la EP que describen los mecanismos patológicos de la enfermedad y su relevancia en el entendimiento de este proceso neurodegenerativo.
Nuevas técnicas para el diseño de modelos transgénicosEstá bien establecido que la EP puede ser de origen esporádico (90-95% del total) o familiar (5-10% del total), por lo que se ha buscado abordar de manera independiente este padecimiento en dos tipos de modelos: los toxicológicos (esporádico) y los genéticos (familiar)19. En el modelado in vitro de la EP se han empleado diversas líneas celulares con el propósito de encontrar candidatos terapéuticos por medio de ensayos farmacológicos y toxicológicos. Dentro de ellas se encuentran las líneas celulares de neuroblastoma SH-SY5Y y feocromocitoma PC12, las cuales representan la degeneración de las neuronas dopaminérgicas (NDA)20,21. Por otra parte están las líneas celulares inmortalizadas como «células humanas mesencefálicas de Lund» (LUHMES), MN9D (ratón), CSM14.1 (rata), y los cultivos primarios posnatales obtenidos de mesencéfalo (ratón y rata)22. A pesar de que estos son buenos modelos para representar la degeneración en las NDA, no poseen el genotipo característico de la EP familiar. Previamente se han generado modelos transgénicos de este tipo mediante transfecciones virales (virus adeno-asociado), aunque la eficiencia de este procedimiento es baja23. En contraste, el empleo de las nuevas tecnologías de ingeniería genética permite obtener modelos transgénicos en tiempos significativamente más cortos además de eficiencias muy elevadas y reducción de costos24.
En esta revisión solo se abordan los modelos de carácter genético, puesto que han evolucionado notablemente debido a la mejora de las herramientas de edición génica, permitiendo asociar genes con funciones específicas de manera más clara, lo que ha posibilitado el diseño de modelos transgénicos más completos que emulen la EP.
Nucleasas con dedos de zincUna de las herramientas para la edición del genoma son las nucleasas con dedos de zinc, enzimas que pueden cortar el ADN de doble hebra en un sitio específico.
Hoy en día se pueden diseñar los dominios de dedos de zinc para dirigirlos a ciertas secuencias del ADN a voluntad, con lo cual las nucleasas de dedos de zinc buscarán secuencias únicas dentro de genomas complejos25.
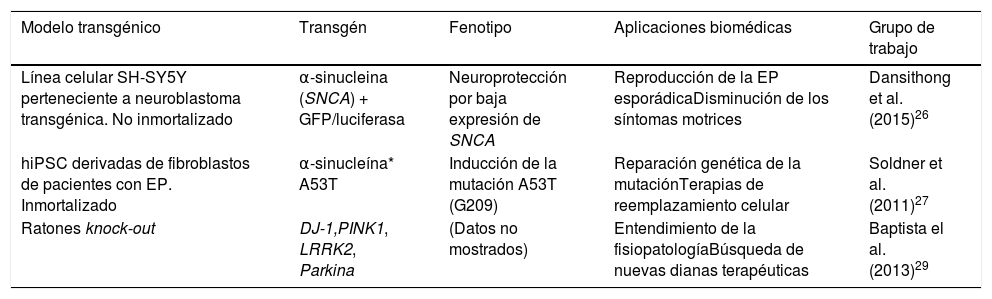
Mediante esta tecnología se han generado modelos celulares transgénicos de la EP, como la creación de líneas celulares que expresan α-sinucleína (SNCA) fusionadas con los genes reporteros de luciferasa o GFP, teniendo como objetivo el probar moléculas que puedan disminuir la expresión de SNCA26. Por otra parte, utilizando este método se ha logrado la reparación genética de la mutación α-sinucleína*A53T en células madre pluripotentes inducidas humanas (hiPSC) derivadas de pacientes con EP27. De igual manera, Sanders et al.28, después de la edición del genoma con ZFN sobre la mutación LRRK2*G2019S en iPSC provenientes de fibroblastos, no detectaron daño mitocondrial en las células progenitoras diferenciadas y neurales. Así mismo, la fundación Michael J. Fox, junto con el Instituto SANGER, han producido varios modelos transgénicos con el fin de recapitular la EP por medio de la tecnología ZFN generando ratas knock-out para los genes DJ-1, PINK1, LRRK2 y Parkina, que exhiben fenotipos distintivos de la EP, útiles para el entendimiento de la fisiopatología y la búsqueda de nuevas dianas terapéuticas y fármacos eficaces29 (tabla 1).
Modelos transgénicos celulares y animales obtenidos por medio de edición con nucleasas con dedos de zinc (ZFN) para enfermedad de Parkinson
| Modelo transgénico | Transgén | Fenotipo | Aplicaciones biomédicas | Grupo de trabajo |
|---|---|---|---|---|
| Línea celular SH-SY5Y perteneciente a neuroblastoma transgénica. No inmortalizado | α-sinucleina (SNCA) + GFP/luciferasa | Neuroprotección por baja expresión de SNCA | Reproducción de la EP esporádicaDisminución de los síntomas motrices | Dansithong et al. (2015)26 |
| hiPSC derivadas de fibroblastos de pacientes con EP. Inmortalizado | α-sinucleína* A53T | Inducción de la mutación A53T (G209) | Reparación genética de la mutaciónTerapias de reemplazamiento celular | Soldner et al. (2011)27 |
| Ratones knock-out | DJ-1,PINK1, LRRK2, Parkina | (Datos no mostrados) | Entendimiento de la fisiopatologíaBúsqueda de nuevas dianas terapéuticas | Baptista el al. (2013)29 |
El desarrollo de nucleasas específicas programables ha proporcionado una herramienta ampliamente aplicable para inducir modificaciones personalizadas sobre las secuencias de genoma diana mediante la nucleasa dimérica FokI.
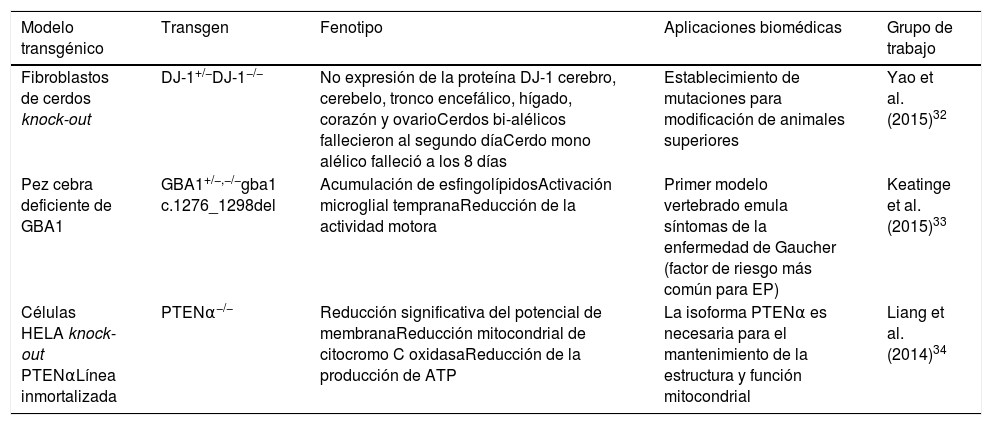
En particular, la tecnología de nucleasas tipo activadores de transcripción (TALEN) es una poderosa herramienta para la ingeniería del genoma, que puede usarse para escindir secuencias genómicas únicas en células vivas y organismos como el pez cebra, la rata y el cerdo30-32. Esta herramienta de edición, en sinergia con el método de transferencia nuclear de células somáticas (SCNT), ha permitido la generación de cerdos knock-out DJ-1 bi-alélicos. El grupo de Yao et al.32 obtuvo un lechón DJ-1+/− y dos lechones DJ-1−/−, en los que posteriormente, en los análisis de secuenciación y western blot, mostraron que la proteína DJ-1 no se expresó en todos los tejidos estudiados (cerebro, cerebelo, tronco encefálico, hígado, corazón y ovario), lo que indica una construcción exitosa del modelo. Otro factor de riesgo involucrado y estudiado en el desarrollo de la EP son las mutaciones de la enzima glucocerebrosidasa1 (GBA1). Para ello, el grupo de Keatinge et al.33 generó, mediante el sistema TALEN, un modelo de pez cebra heterocigoto (GBA+/−) deficiente de GBA1, donde observaron una marcada acumulación de esfingolípidos a los 5días posfecundación acompañado de activación microglial temprana, así como una degeneración pronunciada de las NDA a partir de la octava semana y una reducción llamativa en la actividad motora hacia las 12semanas. Por otra parte, se ha demostrado que la proteína PTEN juega un papel crítico en el mantenimiento de la homeostasis mitocondrial, asociado con el buen funcionamiento neuronal. Recientemente el grupo de Liang et al.34 ha identificado la isoforma PTENα de la proteína PTEN. Mediante el uso de la tecnología TALEN han generado células HELA Knock-out PTENα−/−, encontrando una reducción significativa sobre el potencial de membrana, la actividad mitocondrial de la enzima citocromo C oxidasa y la producción de ATP, demostrando que la isoforma PTENα es necesaria para el mantenimiento de la estructura y de la función de la mitocondria (tabla 2).
Modelos transgénicos celulares y animales generados por nucleasas tipo activadores de transcripción (TALEN)
| Modelo transgénico | Transgen | Fenotipo | Aplicaciones biomédicas | Grupo de trabajo |
|---|---|---|---|---|
| Fibroblastos de cerdos knock-out | DJ-1+/−DJ-1−/− | No expresión de la proteína DJ-1 cerebro, cerebelo, tronco encefálico, hígado, corazón y ovarioCerdos bi-alélicos fallecieron al segundo díaCerdo mono alélico falleció a los 8 días | Establecimiento de mutaciones para modificación de animales superiores | Yao et al. (2015)32 |
| Pez cebra deficiente de GBA1 | GBA1+/−,−/−gba1 c.1276_1298del | Acumulación de esfingolípidosActivación microglial tempranaReducción de la actividad motora | Primer modelo vertebrado emula síntomas de la enfermedad de Gaucher (factor de riesgo más común para EP) | Keatinge et al. (2015)33 |
| Células HELA knock-out PTENαLínea inmortalizada | PTENα−/− | Reducción significativa del potencial de membranaReducción mitocondrial de citocromo C oxidasaReducción de la producción de ATP | La isoforma PTENα es necesaria para el mantenimiento de la estructura y función mitocondrial | Liang et al. (2014)34 |
La reprogramación de células somáticas diferenciadas a células pluripotentes mediante la co-expresión ectópica de factores transcripcionales (Oct4, Sox2, Klf4 y v-Myc) genera las iPSC35. Una vez reprogramadas, son capaces de renovarse y diferenciarse a diversas estirpes celulares, incluyendo las neuronas36. Consecuentemente, las iPSC surgen como un modelo idóneo para la simulación in vitro de diversos procesos patológicos, incluyendo las enfermedades neurodegenerativas como la EP, con el propósito de generar tratamientos novedosos y eficaces como las terapias de reemplazo celular37.
Hoy en día se pueden obtener iPSC de diversos órganos y tejidos, como las células del estómago, hígado, células troncales neuronales, células de sangre periférica, queratinocitos de los folículos del cabello, y especialmente para el estudio de enfermedades neurodegenerativas se obtienen fibroblastos a partir de biopsias de pacientes diagnosticados para su posterior reprogramación a neuronas y corrección con enzimas tipo nucleasas para evaluar su funcionalidad38,39.
Otro método de edición génica es el revolucionario sistema de nucleasas guiadas derivadas de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas en conjunto con la proteína asociada 9 (CRISPR/Cas9), guiado por RNAs hacia el ADN blanco40. Este sistema originalmente pertenece a un mecanismo adaptativo de defensa en procariotas descubierto por primera vez en E.coli41. El sistema CRISPR se encuentra de manera no codificante, para posteriormente transcribirse en repeticiones directas de crRNAs (CRISPR RNA) cortos que contienen secuencias separadoras individuales (20-50pb)42 que direccionan a la nucleasa Cas9 a secuencias específicas de ADN genómico, donde río arriba debe encontrarse una secuencia «motivo adyacente protoespaciador» (PAM) de reconocimiento, la cual es NGG y se encuentra presente en un promedio de 8pb en el genoma humano43. La nucleasa Cas9 corta entonces ambas hebras de ADN con precisión y el daño se repara a través de los mecanismos NHEJ (recombinación no homóloga/unión de extremos no homólogo) y HDR (reparación homóloga dirigida), dando lugar de este modo a interrupciones de genes e inactivación del gen diana. En 2012 Jinek et al.44 fusionaron un crRNA que contenía la secuencia guía de orientación a un tracrRNA, denominado ARN de guía simple (sgRNA), para facilitar la escisión de ADN por CRISPR/Cas9. Asimismo se simplificó el sistema teniendo un solo guía de RNA quimérico (sgRNA), obteniendo de esta manera simplificada una guía para Cas9 sgRNA=crRNA-tracrRNA, el cual es de 20nt, cifra máxima que da la especificidad de reconocimiento del gen de interés que será editado. Este sistema ha sido aprovechado para facilitar manipulaciones genéticas en prácticamente todas las variedades de células y organismos, permitiendo generar organismos con mutaciones múltiples o hacer grandes deleciones cromosómicas45.
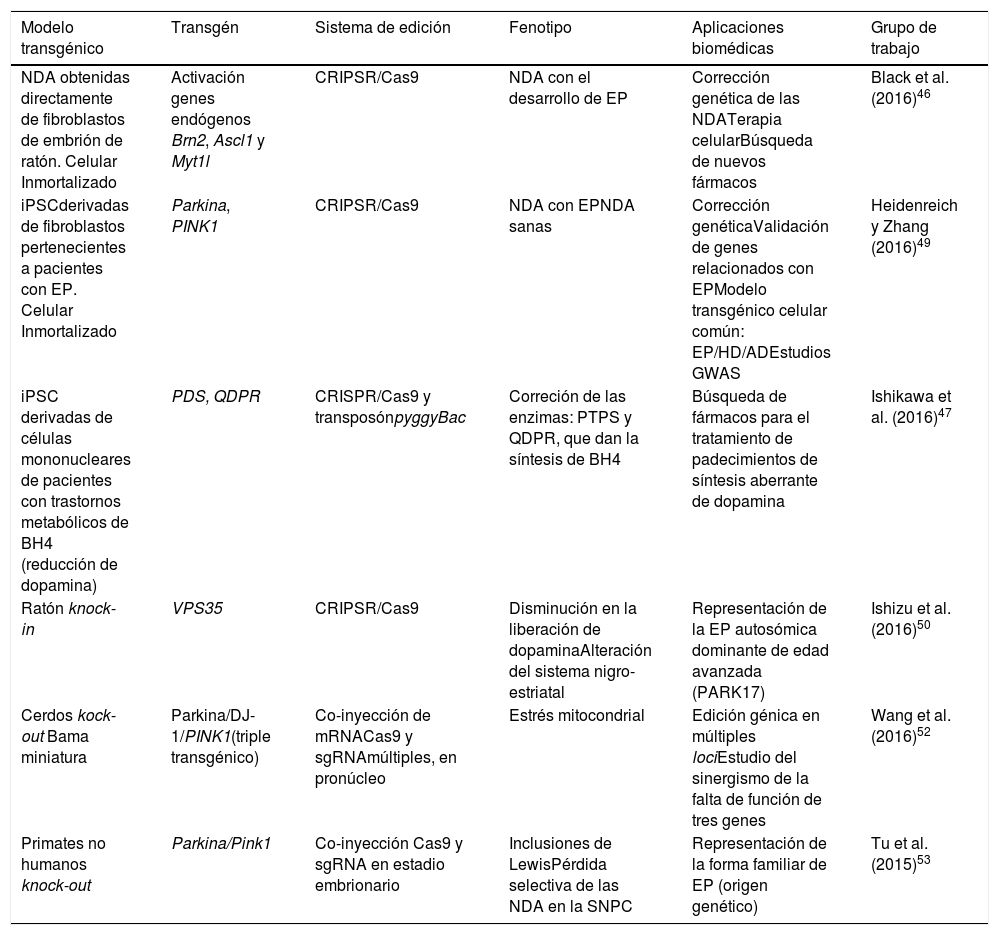
Combinación de tecnologías: células pluripotentes inducidas y el sistema de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadasCon base en lo anteriormente mencionado, diversos grupos de trabajo han comenzado a emplear el sistema CRISPR/Cas9 para la obtención de iPSC con el propósito de modelar la EP. Una de las aplicaciones novedosas de CRISPR es la mejora de la eficiencia de reprogramación directa de fibroblastos a neuronas. Recientemente el grupo de Black et al.46 utilizó CRIPSR/Cas9 para convertir directamente fibroblastos de embrión de ratón en neuronas por medio de la activación de genes endógenos (Brn2, Ascl1 y Myt1l) a nivel epigenético del locus blanco, esto induciendo una expresión endógena que se sostiene durante la reprogramación. La activación y remodelación epigenética de la cromatina en su forma nativa facilita la reprogramación celular y evita el forzamiento de la célula causado por la integración al genoma de plásmidos como los métodos tradicionales de sobreexpresión ectópica, ofreciendo una nueva alternativa para superar las barreras epigenéticas que se presentan en el direccionamiento celular. Otros estudios se están enfocando de manera robusta en la corrección a nivel genómico de las iPSC por medio del sistema CRISPR. Se ha observado que si existe deficiencia de la enzima tetrahidrobiopterina (BH4) los niveles de dopamina se reducen significativamente, asociando el proceso con la EP. El grupo de Ishikawa et al.47, mediante el empleo de CRISPR/Cas9 y el transposón pyggyBac, lograron la corrección genética de las enzimas que codifican para la síntesis de tetrahidrobiopterina (BH4) en iPSC derivadas de células mononucleares de sangre periférica provenientes de pacientes con deficiencia en las enzimas 6-pirovuil-tetrahidropterina sintasa y dihidropteridina reductasa. Posteriormente a la diferenciación de las iPSC a NDA, se observó que la corrección de los genes restauró la cantidad de BH4, los niveles de tirosina hidroxilasa y la producción extracelular de dopamina.
Otra de las aplicaciones de alto impacto de CRISPR/Cas9 en iPSC es en los denominados estudios de asociación del genoma completo (GWAS), donde se evalúan polimorfismos que cambian en un solo par de bases en el ADN (SNPs) y se atribuyen como posibles causantes de una enfermedad (en este caso la EP) en una población masiva48. Un estudio realizado por Heidenreich y Zhang49, basado en GWAS, consistió en tres secciones, partiendo de la obtención de iPSC a partir de fibroblastos de individuos saludables y de pacientes con EP y su posterior diferenciación in vitro a neuronas. 1)En las iPSC derivadas de pacientes enfermos se realiza una edición por medio de CRISPR/Cas9 del sitio-específico de recombinación homóloga (HR) de los genes que se piensa podrían estar involucrados en la patogénesis (genes candidato). Si hay una recuperación del fenotipo y esta es similar a lo mostrado por las neuronas derivadas de pacientes sanos después de la edición, hay una validación del gen y se confirma como gen candidato. 2)Asimismo, en las neuronas derivadas de pacientes sanos se mutan los genes que son presuntos candidatos mediante CRISPR/Cas9, y si existe una recapitulación de la patología (EP) in vitro, se confirman. En base a esto, 3)se promueve la asociación de genes con loci múltiples en común para enfermedades como EP y Alzheimer. Por medio del sistema CRISPR se puede llegar a concretar correlaciones entre fenotipos funcionales y distintas mutaciones genéticas con la aplicación de RNA-guías (sgRNA). Con esta información se podría llegar a establecer próximamente modelos transgénicos celulares con loci en común para distintas enfermedades neurodegenerativas como son la EP, el Alzheimer (AD) y la enfermedad de Huntington (HD), pudiendo derivar estudios de búsqueda de nuevas moléculas asociadas a la patogénesis, así como terapias múltiples funcionales para los tres padecimientos (EP/AD/HD) (tabla 3).
Modelos transgénicos celulares y animales generados por medio del sistema de edición CRISPR/Cas9 para EP
| Modelo transgénico | Transgén | Sistema de edición | Fenotipo | Aplicaciones biomédicas | Grupo de trabajo |
|---|---|---|---|---|---|
| NDA obtenidas directamente de fibroblastos de embrión de ratón. Celular Inmortalizado | Activación genes endógenos Brn2, Ascl1 y Myt1l | CRIPSR/Cas9 | NDA con el desarrollo de EP | Corrección genética de las NDATerapia celularBúsqueda de nuevos fármacos | Black et al. (2016)46 |
| iPSCderivadas de fibroblastos pertenecientes a pacientes con EP. Celular Inmortalizado | Parkina, PINK1 | CRIPSR/Cas9 | NDA con EPNDA sanas | Corrección genéticaValidación de genes relacionados con EPModelo transgénico celular común: EP/HD/ADEstudios GWAS | Heidenreich y Zhang (2016)49 |
| iPSC derivadas de células mononucleares de pacientes con trastornos metabólicos de BH4 (reducción de dopamina) | PDS, QDPR | CRISPR/Cas9 y transposónpyggyBac | Correción de las enzimas: PTPS y QDPR, que dan la síntesis de BH4 | Búsqueda de fármacos para el tratamiento de padecimientos de síntesis aberrante de dopamina | Ishikawa et al. (2016)47 |
| Ratón knock-in | VPS35 | CRIPSR/Cas9 | Disminución en la liberación de dopaminaAlteración del sistema nigro-estriatal | Representación de la EP autosómica dominante de edad avanzada (PARK17) | Ishizu et al. (2016)50 |
| Cerdos kock-out Bama miniatura | Parkina/DJ-1/PINK1(triple transgénico) | Co-inyección de mRNACas9 y sgRNAmúltiples, en pronúcleo | Estrés mitocondrial | Edición génica en múltiples lociEstudio del sinergismo de la falta de función de tres genes | Wang et al. (2016)52 |
| Primates no humanos knock-out | Parkina/Pink1 | Co-inyección Cas9 y sgRNA en estadio embrionario | Inclusiones de LewisPérdida selectiva de las NDA en la SNPC | Representación de la forma familiar de EP (origen genético) | Tu et al. (2015)53 |
Mediante esta tecnología se han creado de manera eficiente modelos animales de EP con la finalidad de comprender el proceso natural de la enfermedad asociado a mutaciones puntuales y el desarrollo de nuevas terapias. Hoy en día, mediante el empleo de la tecnología de CRISPR/Cas9 se han generado modelos knock-in, como el ratón transgénico Vps35 D620N, para estudiar la patogénesis de PARK17 en la EP de aparición tardía asociada a una mutación puntual en el gen35, obteniéndose ratones transgénicos que expresan la proteína homóloga mutante y no observando muerte prematura o neurodegeneración clara, hasta las 70 semanas, en los ratones heterocigotos y homocigotos50. Por otra parte se han estudiado los genes implicados en la EP autosómica recesiva PARK2 y PINK1 en un modelo de cerdo doméstico utilizando el sistema CRISPR/Cas9 combinado con el método SCNT, y primeramente se obtuvieron con éxito homocigotos dobles knock-out para los genes PARK2 y PINK1, con una eficiencia aproximada del 38%. Los cerdos transgénicos nacidos y viables no presentaron durante 7meses de observación síntomas típicos de la enfermedad, como agitación, rigidez, lentitud de movimiento y dificultad para caminar51. Recientemente Wang et al.52 obtuvieron un modelo humano de la EP (knock-out) en embriones pronucleares obtenidos de cerdos Bama miniatura, por medio de co-inyección in vivo de mRNACas9 y sgRNA múltiples dirigida a tres loci: Parkina/DJ-1/PINK1. Exhibieron una robusta capacidad de edición múltiple en la producción de un modelo transgénico con eficiencia del 100%, sin la inducción de mutaciones en sitios inespecíficos significativos (off-target). Los cerdos no mostraron síntomas característicos de la EP hasta después de los 10meses de edad, atribuyéndose un sinergismo producido por la inactivación efectiva de los tres loci. La facilidad de edición genética múltiple por medio de CRISPR/Cas9 en cerdos ofrece gran valor en el área médica y agrícola venidera.
Por otro lado, se puede llevar a cabo la edición de estos genes mediante CRISPR/Cas9 en primates no humanos y en otros animales grandes, inactivando la expresión génica. Cuando ambos alelos están mutados (null−/−) se produce una pérdida total de la funcionalidad de Parkina y Pink1, teniendo como consecuencia un modelo que representa la forma familiar de la EP. Para asegurar que hay una interrupción en la expresión de los alelos se pueden diseñar múltiples regiones blanco que pueden ser monitoreadas mediante la co-inyección de RNA guías (sgRNA) y Cas9 en óvulos fertilizados en el estadio de embrión (una sola célula)53. De esta manera los primates no humanos transgénicos exhiben síntomas motrices complejos como bradicinesia, temblor, rigidez, inestabilidad postural, además de inclusiones de cuerpos de Lewis, característicos de los pacientes con EP54 (fig. 1 y tabla 3).
 Selección de genes candidatos para EP por medio de CRISPR/Cas9 en neuronas diferenciadas de iPSC obtenidas de fibroblastos de pacientes mediante los estudios de asociación del genoma completo (GWAS) y sus implicaciones futuras44 (modificado de Heidenreich y Zhang). 2) Reprogramación directa de fibroblastos a NDA por medio de manipulación epigenética con CRISPR/Cas9. No existe un sobreesfuerzo celular debido a que no hay sobreexpresión ectópica, por lo tanto el direccionamiento a diferentes estirpes celulares es más sencillo. 3) Generación de modelos transgénicos animales por edición de genes a nivel embrionario, asociados con EP del tipo familiar. Se puede reproducir la fisiopatología típica, así como las manifestaciones conductuales características de la EP.")
Modelos transgénicos de EP generados con CRISPR/Cas9.
1) Selección de genes candidatos para EP por medio de CRISPR/Cas9 en neuronas diferenciadas de iPSC obtenidas de fibroblastos de pacientes mediante los estudios de asociación del genoma completo (GWAS) y sus implicaciones futuras44 (modificado de Heidenreich y Zhang).
2) Reprogramación directa de fibroblastos a NDA por medio de manipulación epigenética con CRISPR/Cas9. No existe un sobreesfuerzo celular debido a que no hay sobreexpresión ectópica, por lo tanto el direccionamiento a diferentes estirpes celulares es más sencillo.
3) Generación de modelos transgénicos animales por edición de genes a nivel embrionario, asociados con EP del tipo familiar. Se puede reproducir la fisiopatología típica, así como las manifestaciones conductuales características de la EP.
Una de las características más fundamentales, como se mencionó previamente, es que el sistema CRISPR/Cas9 tiene la facultad de generar mutaciones básicamente en cualquier lugar del ADN genómico55-57. Esta ventaja es primordial para la generación de transgénicos en animales superiores para modelar la EP, puesto que la co-inyección de Cas9 y sgRNA simplifica altamente la generación de las mutaciones de interés, tópico que mostraba enormes inconvenientes en años anteriores. Horii et al.58 evaluaron tres técnicas de microinyección mediante las cuales se pueden generar transgénicos a nivel embrionario en ratones: la entrega de ADN en el pronúcleo, de RNA en el pronúcleo y de RNA en el citoplasma, concluyendo que la inyección de RNA en el citoplasma es la más eficiente en el estadio de blastocisto y en la producción de crías, mostrando adyacentemente que la inyección de ADN y ARN en el pronúcleo es perjudicial para la viabilidad de los embriones. Esto es un factor trascendental a considerar si se quiere generar modelos transgénicos de ratón funcionales por medio de CRISPR/Cas9 a nivel embrión.
Además de los modelos transgénicos generados por medio de nucleasas, otros modelos recientes se enfocan específicamente en la alteración de factores de transcripción, esenciales para el desarrollo de las NDA maduras (NDAm), promoviendo la baja celular en la SNPC. Los modelos más relevantes se discuten a continuación.
Modelos transgénicos de la enfermedad de Parkinson derivados de factores de transcripción necesarios para el desarrollo de neuronas dopaminérgicasOtro sector emergente en el modelado de la EP es el desarrollo de ratones con mutaciones dirigidas a factores de transcripción importantes en la generación, desarrollo y mantenimiento de las NDA adultas (fig. 2). Diversos estudios han llevado al desentrañamiento de la genética y nuevas vías de señalización de los procesos que controlan la generación de las NDA maduras59, presentando la expresión de Engrailed-1/Engrailed-2 (En−/−), Foxa1/2, Nurr1, Otx2 y Pitx3 (entre otros) para el mantenimiento de las NDA de mesencéfalo (NDAm) de por vida60,61. Dentro de los ejemplos más relevantes se ha demostrado la asociación de estos factores de transcripción con funciones neuroprotectoras en la EP; por ejemplo, la expresión de Otx2 previene la pérdida de NDAm en ratones Engrailed-1 heterocigotos En1(+/−)62. Se ha visto que la homeoproteína «Engrailed» interviene en la activación de la vía de mTOR (vía esencial en la célula) y la regulación de la autofagia63, además confiere protección a las NDAm frente a la exposición a MPTP, por medio de la traducción de las subunidades del Subconjunto complejo I mitocondrial, mejorando su actividad e incrementando la síntesis de ATP64,65. Esto indica la importancia de la traducción de RNAm correspondientes a componentes de la mitocondria, puesto que muchos estudios sustentan que la falla de suministro de la elevada energía que requieren las NDAm está directamente relacionada con gran parte de la patogénesis de la EP66,67. Otro ratón knock-out de esta índole se realiza mediante la modificación de Nurr1, el cual está regulado por α-sinucleína mutada68, y se piensa que esto puede comprometer la señalización GDNF/Ret (factor neurotrófico de línea celular-derivada de glía), cuya función se atribuye a la sobrevivencia de las NDA (entre otros tipos de neuronas) y se ha propuesto como posible blanco terapéutico para la EP69. Un estudio reciente demostró que la expresión forzada de Nurr1 y el factor de transcripción Foxa2 (crucial para la especificación y diferenciación de las NDA) en células de la glía tienen un efecto protector para las NDAm en un modelo murino de EP con exposición a MPTP70,71. El mecanismo de acción observado fue sinérgico para Nurr1 y Foxa2 en microglía, mediante la disminución de citocinas proinflamatorias y el incremento de la síntesis de factores neurotróficos como GDNF, BDNF, NT3, SHH (entre otros), los cuales regulan los mecanismos de recuperación, regeneración y protección naturales del tejido nervioso70.
, Foxa1/2, Nurr1, Pitx3 y PGC-1α, así como las vías de señalización en las que intervienen y la fisiopatología producida por los knock-out (fenotipo).")
Modelos transgénicos murinos asociados a factores de transcripción. Los factores de transcripción mutados en estos modelos son esenciales para la generación, desarrollo y mantenimiento de las NDA adultas. Se describen esquemáticamente los modelos generados en las mutaciones de los factores de transcripción: Engrailed-1/Engrailed-2 (En−/−), Foxa1/2, Nurr1, Pitx3 y PGC-1α, así como las vías de señalización en las que intervienen y la fisiopatología producida por los knock-out (fenotipo).
Pitx3 es otro factor que está muy involucrado en vías de supervivencia a largo plazo y mantenimiento de las NDAm. Se generó un ratón nulo para Pitx3 denominado Aphakia, observando un decremento de la cantidad de NDA en la substancia nigra y en la expresión de tirosina hidroxilasa en el estriado72. Aunadamente Pitx3 tiene como blanco Aldh1a1 (gen codificante para la enzima aldehído deshidrogenasa), indispensable en la producción de ácido retinoico y promotor de actividades antiapoptóticas y antioxidantes en subpoblaciones de NDAm73, por lo que este modelo se puede emplear para propiciar una pérdida selectiva de las NDAm, escenario idóneo de estudio en un modelo de EP.
Diversos estudios han encontrado una asociación entre el receptor co-activador transcripcional del proliferador de peroxisomas-γ (PGC-1α) y la EP en modelos tanto animales como celulares. El grupo de Jiang et al.74 demostró en ratones knock-out de PGC-1α la pérdida significativa de NDA, acompañada de un reducción de dopamina en el cuerpo estriado, mostrando una relación directa entre la pérdida de PGC-1α y la baja celular de las NDA en la SNPC. Este hallazgo es un claro ejemplo de las grandes implicaciones de la realización de modelos transgénicos adecuados al establecer una relación directa entre knock-out y fenotipo asociado a pérdida de función. Aunado a esto, es relevante mencionar que los trabajos previamente descritos de los modelos murinos basados en factores de transcripción no reportan el empleo del sistema CRISPR/Cas9, ni de otras nucleasas, por lo que sería interesante la aportación de estas tecnologías a la facilitación del diseño de los modelos transgénicos.
Modelos de la enfermedad de Parkinson derivados con nucleasas relacionados con el envejecimientoEstá bien establecido, que el envejecimiento es una característica de enorme peso en muchas enfermedades neurodegenerativas y se posiciona como el principal factor de riesgo para desarrollar EP75,76. Según una revisión exhaustiva realizada por Reeve et al.77, existen cambios relacionados con la edad y el envejecimiento que propician un ambiente hostil en la substancia nigra, promoviendo la muerte de las NDA. Dentro de esos procesos se describe al metabolismo de la dopamina, la acumulación de hierro, la conducción del calcio, las mutaciones mitocondriales (mtDNA), la acumulación de neuromelanina y una disminución en la eficiencia de degradación de las proteínas. Se ha observado en diversos trabajos que estos cambios son suficientes para la pérdida de neuronas del sistema nervioso por sí solos, conduciendo en algunos casos a la EP. Una estrategia experimental para mimetizar el envejecimiento en modelos in vitro se describe en el trabajo de Studer et al.78, donde postulan la inducción de parámetros relacionados con la edad en ambas poblaciones celulares, tanto sanas como enfermas, sin la inducción de toxicidad. Estos paradigmas pueden funcionar como una herramienta robusta para la medición de los cambios relacionados con la edad, contemplando la interacción de la genética (líneas isogénicas) con el ambiente en fenotipos de la enfermedad. Una de las opciones novedosas de la generación de modelos que emulen el envejecimiento es la aplicación de las nuevas herramientas de edición basadas en nucleasas para la manipulación directa de las características celulares y moleculares clave, relacionadas con la edad. El sistema CRISPR/Cas9 facilitará enormemente el flanqueo de vías de señalización o la combinación de las mismas. Un ejemplo reciente se describe en el trabajo publicado por el grupo de investigación de Harel et al.79, que realizaron la manipulación de marcadores del envejecimiento referentes al acortamiento de telómeros, alteraciones epigenéticas, pérdida de la proteóstasis y el sentido de la nutrición (genes TERT, ASH2L, ATG5, IGF1R, RPS6KB1) en el pez africano turquoise killifish, de vida-corta, estableciendo una línea de producción de nuevos modelos de vertebrados con una escala de envejecimiento reducida (6meses) propicios para la búsqueda de blancos terapéuticos más fiables relacionados con los signos de la edad. Esto es un avance trascendental, debido que la instauración de un modelo que asemeje el envejecimiento y la neurodegeneración es compleja, partiendo de la premisa de que la gran mayoría de los modelos animales desarrollan la sintomatología típica y mueren jóvenes, cuando las personas con trastornos relacionados a la edad desarrollan los síntomas en la vejez y mueren cuando tienen una edad avanzada, convirtiéndose en un tremendo problema a la hora de extrapolar resultados a la clínica80-82.
Es, por tanto, un área emergente donde es necesario la aplicación de nucleasas (ZFN, TALEN y CRISPR/Cas9) para generar líneas de producción con métodos de alto rendimiento (high-throughput methods) para así develar nuevos blancos terapéuticos relacionados con las características clave que se ven afectadas en los procesos neurodegenerativos relacionados con el envejecimiento.
Desventajas del empleo de nucleasas para la generación de modelos transgénicosNucleasas con dedos de zincLas nucleasas con dedos de zinc (ZFN) formaron parte de la primera generación de herramientas para la edición génica dirigida y se han empleado desde la generación de modelos de patologías en animales hasta la implementación de terapias en humanos83. Sin embargo, se han reportado severas desventajas en su uso experimental con respecto a las otras nucleasas. Dentro de ellas, la más trascendental comprende a la ingeniería de la herramienta misma, debido a que se ha probado que no es fácil el proceso de ensamblaje de los dominios de dedos de zinc con una secuencia determinada de nucleótidos84. Otro inconveniente observado es el limitado acceso que presenta para el reconocimiento del sitio blanco de interés. Se ha demostrado que los ZFN tienen reconocimiento de sitios de unión cada 200 pares de bases (pb) en el DNA genómico, y esto ha podido reducirse hasta 50pb en los dedos de zinc comerciales85. El proceso de generación de modelos knock-out no se ve afectado, debido a que la edición con ZFN conlleva a un corrimiento del marco de lectura en la secuencia codificante temprana, llevando a cabo pequeñas inserciones y deleciones (indel) al azar en cualquier parte del gen, promoviendo la pérdida de función. Contrario a ello, hacer modificaciones en una secuencia específica se torna bastante complejo, haciendo ineficiente esta herramienta para la incorporación de sustituciones o reemplazos de secuencias en el genoma (knock-in)86-90.
Nucleasas tipo activadores de transcripciónEsta tecnología surgió dos años antes que el sistema CRISPR, y en teoría se puede realizar el rompimiento de doble cadena del DNA en cualquier parte del genoma, con la presencia de sitios de reconocimiento conocidos de unión a dominios de DNA utilizando nucleasas TALEN artificiales91. Una de las limitaciones pertinentes que presenta esta tecnología es que requiere la presencia del nucleótidoT (timina) antes del término5’ de la secuencia blanco. Para la selección del sitio de unión se debe considerar una variación en la longitud de la secuencia espaciadora. Se ha demostrado que la interacción del residuo W232 en la región N-terminal del dominio de unión a DNA con la timina-N-terminal afecta la eficiencia de unión del complejo TALEN al sitio blanco92. Sin embargo, estas complicaciones pueden ser sobrellevadas mediante la producción y selección de mutantes distintos que posean dominios TALEN-N terminal que sean propensos a unión a los nucleótidos A, G o C.
El sistema de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadasPese a las múltiples bondades que ofrece el sistema CRISPR, se han identificado varios parámetros inherentes al mecanismo de acción de esta nucleasa, que influyen directamente sobre la disminución de la eficiencia de edición. Se describen dentro de ellos: el diseño adecuado de los RNA guías (sgRNAs) que actuarán sobre el sitio blanco, los métodos de entrega del material genético al núcleo, la actividad de la enzima Cas9, los efectos off-target y la baja incidencia del mecanismo de reparación homóloga (HDR) en la células; asimismo, se ha observado un impacto directo en la especificad, la eficiencia y la fidelidad del sistema93.
El diseño inadecuado de los sgRNA compromete la especificidad y la eficiencia de la edición dirigida94. Para propiciar un diseño experimental robusto se requiere la generación de diversos sgRNA, puesto que se ha observado en múltiples experimentos que algunos de ellos pueden ser menos eficientes o también inactivos94-98. Asimismo se ha observado que el orden y la composición de los sgRNAs favorecen o afectan el proceso de edición. Por ejemplo, en el extremo terminal 5’ de los sgRNAs es necesaria la presencia del nucleótidoG (guanina) para la expresión del promotor U694. También se recomienda que G se encuentre en primera o segunda posición con respecto a la secuencia PAM, donde la enzima Cas9 se unirá a la secuencia selectivamente. Una colocación de C (citocina) en las posiciones mencionadas será desfavorable para el proceso. Debido a una baja expresión de sgRNAs cuando hay múltiples U (uracilo), no es recomendable la presencia de múltiples T (timina) en posiciones adyacentes a PAM95. Cuando los sgRNAs son ricos en G y tienen una cantidad pobre de A (adenina), se incrementa la estabilidad y la eficiencia96. Otro factor relevante en la especificidad es la generación de efectos off-target. A pesar de que el sistema CRISPR/Cas9 tiene mayor especificidad que las otras nucleasas, tanto en modelos in vitro como in vivo, se ha reportado la producción de múltiples mutaciones en sitios inespecíficos, lo que en estudios biológicos, terapia génica y la edición dirigida son efectos sumamente indeseables. Distintos estudios reportan que el rompimiento de DNA de doble cadena mediado por Cas9 puede ser inhibido por la pérdida del reconocimiento único en los nucleótidos de la secuencia (mismatch) en la región complementaria del sgRNA, con la secuencia blanco, especialmente en la región nucleotídica contigua a PAM99,100. Otros trabajos basados en secuenciación de todo el genoma (whole-genome sequencing) evidencian que la presencia de mismatches en la región terminal de los sgRNAs es mejor tolerada99,100. Sin embargo, en células humanas se ha visto que por encima de 5 mismatches se puede dar el corte de secuencias no blanco, y en esos sitios se dan mutaciones en frecuencias a un nivel similar a las que se predeterminaron como blanco on-target101.
Como se mencionó previamente, el mecanismo de acción de la enzima Cas9 aprovecha las vías naturales de reparación de la célula, la de unión de extremos no-homólogos (NHEJ) y la de recombinación homóloga (HDR). Al realizar el corte en el sitio blanco se reclutan diferentes proteínas que llevarán a cabo la reparación del daño; para el mecanismo NHEJ: KU70, DNA-PKcs, Artemis, XRCC4, pol μ, XLF/Cernunnos, KU80 y ligasaIV102, mientas que para HDR: RAD50, RAD51, MRE 11, Nbs1, XRCC2, XRCC3, RAD52, RAD54B, BRCA2102 y la invasión por un templado de DNA donador homólogo103-105. Sin embargo, la proporción en la que ocurren estos dos mecanismos es desigual; el mecanismo NHEJ es más probable que el de HDR, siendo NHEJ un mecanismo burdo que no permite realizar sustituciones nucleotídicas ni una edición precisa93. Es por eso que el incremento de la incidencia del mecanismo HDR es un desafío que potenciará la eficiencia en el sistema CRISPR/Cas9, conllevando a una mejoría en el ámbito de terapia génica y la generación de modelos transgénicos.
ConclusiónUno de los principales inconvenientes que presenta la EP es la falta de modelos que emulen la fisiopatología causada por la neurodegeneración de las NDA y la proteinopatía, teniendo por consecuencia tratamientos inadecuados e ineficaces. Por tanto, es trascendental recrear lo más precisamente posible las características típicas de este padecimiento en un modelo transgénico mediante el empleo de herramientas de edición génica basadas en nucleasas: ZFN, TALEN y CRISPR/Cas9, lo que representa una oportunidad para la generación de modelos que mimeticen los síntomas característicos, para una mejor comprensión de la etiología y los mecanismos moleculares involucrados en el proceso de iniciación y desarrollo de la EP. Esto en aras de poder realizar estudios novedosos de diagnóstico temprano, la búsqueda de potenciales dianas terapéuticas, la corrección selectiva de genes o mutaciones causales de la enfermedad y la selección de moléculas que puedan detener o ralentizar la progresión de la enfermedad. Por otra parte, las nuevas tecnologías de reprogramación de las iPSC desabren alternativas ampliamente favorables para la superación de las barreras presentes en el direccionamiento celular, propiciando el futuro establecimiento de la medicina personalizada, la búsqueda efectiva de fármacos novedosos y el desarrollo de terapias de reemplazo celular aplicadas en la clínica, no solo para la EP, sino también para otros padecimientos complejos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Proyecto Fondo CONACyT 2016, Apoyo al Fortalecimiento y Desarrollo de la Infraestructura Científica y Tecnológica, No. 271307, y Red Temática de Células Troncales y Medicina Regenerativa del CONACyT.

