



La esclerosis múltiple (EM) es una enfermedad autoinmune desmielinizante del sistema nervioso central (SNC) en la que los astrocitos tienen una participación importante como células inmunes del SNC, aunque su actividad como células presentadoras de antígeno (APC) aún es discutida.
DesarrolloEn la presente revisión se analiza la evidencia existente sobre la participación de los astrocitos en la inflamación del SNC en la EM, así como diversos mecanismos que modifican su actividad en la enfermedad.
ConclusionesLos astrocitos desempeñan un papel trascendental en la patogénesis de la EM, debido a que expresan receptores TLR, así como proteínas del complejo principal de histocompatibilidad (MHC) clasei yii. Además, participan en la regulación de la barrera hematoencefálica (BHE) y la modulación de la actividad de los linfocitosT mediante la producción de citocinas. Futuros estudios deberán enfocarse en el papel de los astrocitos con el objetivo de encontrar nuevos blancos terapéuticos para el tratamiento de la EM.
Multiple sclerosis (MS) is a demyelinating autoimmune disease of the central nervous system (CNS), in which astrocytes play an important role as CNS immune cells. However, the activity of astrocytes as antigen-presenting cells (APC) continues to be subject to debate.
DevelopmentThis review analyses the existing evidence on the participation of astrocytes in CNS inflammation in MS and on several mechanisms that modify astrocyte activity in the disease.
ConclusionsAstrocytes play a crucial role in the pathogenesis of MS because they express toll-like receptors (TLR) and major histocompatibility complex (MHC) classI andII. In addition, astrocytes participate in regulating the blood-brain barrier (BBB) and in modulating T cell activity through the production of cytokines. Future studies should focus on the role of astrocytes in order to find new therapeutic targets for the treatment of MS.
La esclerosis múltiple (EM) es una enfermedad autoinmune desmielinizante del sistema nervioso central (SNC)1-3 con una gran heterogeneidad en su evolución clínica4; está asociada con diversos factores de riesgo, tanto genéticos como ambientales5. La principal característica de esta enfermedad es la aparición de lesiones o placas6 formadas por células residentes del SNC y células del sistema inmune (SI), las cuales migran a través de la barrera hematoencefálica (BHE) e inducen procesos inflamatorios en el SNC7. Se conoce que los linfocitosT y los macrófagos que invaden el SNC son las principales células que dirigen la inflamación del SNC y modulan la actividad de la glía y las neuronas a través de diversos mecanismos celulares que conducen a la desmielinización y muerte neuronal en los pacientes con EM8.
Los astrocitos son las células gliales más abundantes del SNC9 y desempeñan un papel dual en la patogénesis de la EM10. Por un lado, se ha descrito que los astrocitos contribuyen a la progresión de la enfermedad a través de su actividad como células inmunes del SNC y por la producción de quimiocinas que facilitan la migración de células inmunes de la sangre periférica al interior del SNC10; por otro lado, los astrocitos promueven la migración, proliferación y diferenciación de células precursoras de oligodendrocitos11, lo que favorece la remielinización en la EM. La evidencia con la que se cuenta actualmente sugiere que es necesario revalorar la posición de los astrocitos, no solo como células de soporte y mantenimiento de la homeostasis, sino como actores principales en las enfermedades autoinmunes del SNC, de forma notable en la EM.
Con base en lo anterior, el objetivo del presente trabajo es revisar, integrar y discutir la evidencia sobre el papel de los astrocitos en la patogénesis de la EM. En esta revisión se analizará lo siguiente: a)la participación de los astrocitos en el proceso inflamatorio de la EM como células inmunes del SNC; b)la función de los astrocitos como células presentadoras de antígeno (antigen presenting cells [APC]); c)la participación de los astrocitos en el mantenimiento de la BHE y algunos mecanismos que modifican su permeabilidad, lo que facilita la diapédesis de las células inmunes al SNC, y d)la participación de los astrocitos en la regulación de los linfocitosT.
Los astrocitos como células inmunes en la esclerosis múltipleLos astrocitos, al igual que la microglía, expresan receptores de reconocimiento de patrón (pattern recognition receptor [PRR]) que se unen a patrones moleculares asociados a patógenos (pathogen-associated molecular patterns [PAMP])12. Dichos PRR reconocen secuencias moleculares de agentes patógenos, como bacterias13,14 y virus15,16, así como patrones moleculares asociados a daño (damage-associated molecular patterns [DAMP]), considerados como «propios» y que son liberados después de un daño o muerte celular17. En el caso de los PRR, los astrocitos expresan constitutivamente receptores de tipo Toll-3 (toll-like receptor [TLR3]) y TLR412. El TLR3 es un receptor que reconoce ARN de doble cadena liberado de células dañadas o células infectadas por virus, lo que induce una respuesta proinflamatoria por el SI18 a través de una señalización dependiente de TRIF, NF-κB, RIP1 y TRAF619. Sin embargo, Bsibsi et al.20 demostraron que en el SNC la activación de los astrocitos a través del TLR3 induce la sobreproducción de las citocinas antiinflamatorias interleucina-9 (IL-9), IL-10 e IL-11, así como una disminución en la expresión de la subunidad p40 de IL-12 e IL-23. Además, la expresión de TLR3 en los astrocitos puede potenciarse mediante el estímulo por interferón-gamma (IFN-γ), IFN-β o IL-1β21. La evidencia anterior sugiere que la expresión de TLR3 en pacientes con EM modula la progresión de la inflamación mediante la expresión de citocinas antiinflamatorias, que limitan la actividad de las células inmunes.
Por otro lado, se ha demostrado que el TLR4 se encuentra sobreexpresado en lesiones cerebrales de animales con encefalomielitis autoinmune experimental (EAE), modelo experimental de la EM, así como en células sanguíneas de pacientes con EM remitente-recurrente (EM-RR) y secundaria-progresiva (EM-SP)22. Crowley et al.23 encontraron que la activación de TLR4 y TLR3 con un estímulo inflamatorio, en pacientes con EM, induce un incremento en la expresión del NF-κB que provoca una elevación de los niveles de TNF-α. Esta evidencia soporta la idea de que los astrocitos desempeñan una función dual en la EM, ya que por un lado la activación de TLR3 en los astrocitos provoca la liberación de citocinas antiinflamatorias que limitan la neuroinflamación, mientras que por el otro, el mismo TLR3 junto con TLR4, tras un estímulo inflamatorio, podrían promover el proceso proinflamatorio, mediante la producción de TNF-α.
El papel de los astrocitos como células presentadoras de antígeno en la esclerosis múltipleLos astrocitos son descritos como APC «no tradicionales»24, porque se considera que solo adquieren funciones inmunes cuando ocurre un daño en el SNC25. Los astrocitos expresan constitutivamente el complejo principal de histocompatibilidad (Major Histocompatibility Complex [MHC]) clasei26. Sin embargo, la expresión de MHC claseii en los astrocitos es controversial, ya que existe evidencia que demuestra su expresión de forma constitutiva27, mientras que en otros estudios se encontró que su expresión es mínima28 o que debe ser inducida por citocinas29. Además, existe controversia sobre la expresión de moléculas coestimuladoras, debido a que algunos estudios afirman que los astrocitos pueden expresar moléculas de la familia B730,31, mientras que otros no encontraron evidencia de la expresión de las mismas32,33. Sin embargo, queda claro que los procesos infecciosos, como las infecciones virales, inducen la expresión de MHC clasei yii en los astrocitos34,35 y que la expresión de ambos MHC puede ser modificada de acuerdo al perfil de citocinas que predomine en el SNC27,36-38.
Estudios realizados en lesiones de biopsias de tejido cerebral post mortem de pacientes con EM han demostrado la presencia MHC clasei39 yii40,41 en astrocitos (fig. 1). Se ha descrito que los linfocitosT presentes en las lesiones de EM inducen la sobreexpresión de MHC clasei mediante la producción de IFN-γ42 y que dicha expresión es regulada por la actividad de NF-κB26. El IFN-γ también incrementa la expresión de MHC claseii27,36,43 a través del incremento de la actividad de la cinasa de proteínaC (protein kinase C [PKC]) y la potenciación de la actividad del factorx (IFN-gamma-enhanced factorX [IFNEX]), que interactúa con la cajax del promotor de DRA44. Además, IFN-γ induce un incremento en la movilidad dependiente de los filamentos intermedios de los compartimentos intracelulares, que contienen moléculas de MHC claseii en astrocitos, lo que modifica la velocidad de transporte de las moléculas de MHC hacia la superficie36. Lo anterior sugiere que los astrocitos podrían expresar constitutivamente MHC claseII, el cual se encuentra en los compartimentos intracelulares hasta que los astrocitos reciban el estímulo de IFN-γ.
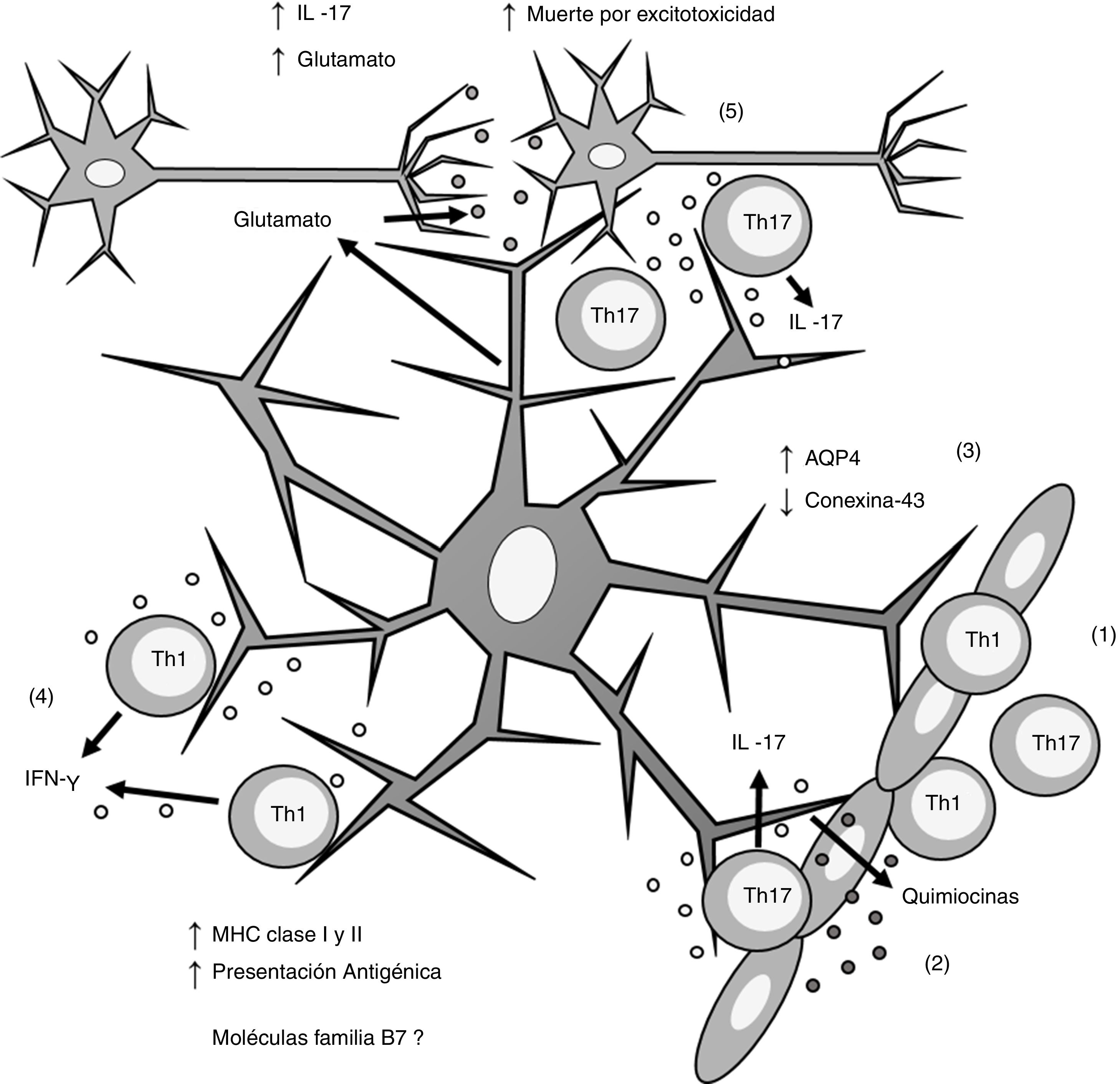
 Los linfocitos Th1 y Th17 migran por diapédesis a través de la barrera hematoencefálica; (2) los linfocitos Th17 producen IL-17 que induce la producción de quimiocinas por los astrocitos, lo que incrementa el reclutamiento de células inmunes; (3) los astrocitos sobreexpresan AQP4 y disminuyen la expresión de conexina-43; (4) los linfocitos Th1 producen IFN-γ que induce la sobreexpresión de MHC clasei yii en los astrocitos, lo que incrementa la presentación antigénica; (5) los linfocitos Th17 producen IL-17 que induce un incremento de glutamato en la sinapsis neuronal, lo que provoca excitotoxicidad y muerte neuronal. AQP4: acuaporina 4; IFN-γ: interferón gamma; IL-17: interleucina 17; MHC: complejo principal de histocompatibilidad; Th1: linfocitos Th1; Th17: linfocitos Th17.")
Esquema general de la participación de los astrocitos en la patogénesis de la esclerosis múltiple. (1) Los linfocitos Th1 y Th17 migran por diapédesis a través de la barrera hematoencefálica; (2) los linfocitos Th17 producen IL-17 que induce la producción de quimiocinas por los astrocitos, lo que incrementa el reclutamiento de células inmunes; (3) los astrocitos sobreexpresan AQP4 y disminuyen la expresión de conexina-43; (4) los linfocitos Th1 producen IFN-γ que induce la sobreexpresión de MHC clasei yii en los astrocitos, lo que incrementa la presentación antigénica; (5) los linfocitos Th17 producen IL-17 que induce un incremento de glutamato en la sinapsis neuronal, lo que provoca excitotoxicidad y muerte neuronal.
AQP4: acuaporina 4; IFN-γ: interferón gamma; IL-17: interleucina 17; MHC: complejo principal de histocompatibilidad; Th1: linfocitos Th1; Th17: linfocitos Th17.
Por el contrario, la expresión de MHC puede ser inhibida en los astrocitos mediante diversas moléculas. Una de las primeras moléculas con este efecto que se estudió fue el IFN-β, el cual bloquea la expresión de MHC claseii previamente inducida por el IFN-γ45,46, sin modificar la expresión de MHC clasei46. Este efecto inmunomodulador es aprovechado como un mecanismo de acción del tratamiento con IFN-β para pacientes con EM47-49, aprobado por la agencia del gobierno de los Estados Unidos de América para la regulación de alimentos y medicamentos (Food and Drug Administration [FDA]) en 199350. Este efecto inmunomodulador favorece un retraso en la aparición de recaídas, la disminución del deterioro neurológico y la disminución de la progresión de la enfermedad50.
Otras citocinas asociadas con la modulación de la expresión de MHC claseii en astrocitos son IL-438, IL-1α27 y TGF-β51. En este sentido, TGF-β es la más estudiada y se conoce que reduce la expresión de MHC claseii en astrocitos51,52 y otros tipos de células53,54. Dong et al.55 demostraron que el TGF-β inhibe la expresión en astrocitos del transactivador del MHC claseii (Class II Transactivator [CIITA]), necesario para la inducción de la EAE56, así como la actividad del promotoriv de CIITA a través del factor de transcripción Smad355.
Existen algunos estudios que refieren la posible regulación de la expresión de MHC mediante moléculas directamente involucradas en la transmisión nerviosa. En esta línea se ha reportado que el glutamato57, la norepinefrina57,58 y los gangliósidos59,60 pueden inhibir la expresión del MHC en astrocitos. Además de los anteriores, se ha descrito que la serotonina puede modificar la respuesta de las células del SI61,62. Se conoce que los agonistas de los receptores de serotonina (HTR) inhiben la expresión de MHC claseii inducida por IFN-γ, así como de moléculas coestimuladoras de la familia B7 en astrocitos63. Seddighzadeh et al.64 encontraron una interacción entre un haplotipo protector en los alelos del receptor de serotonina HTR2A y HLA-DRB1 expresados constitutivamente en fibroblastos de tejido sinovial en pacientes con artritis reumatoide, una enfermedad autoinmune ampliamente estudiada. Debido a que los astrocitos se encuentran en la formación de la sinapsis nerviosa65, las modificaciones en la liberación de neurotransmisores podrían modificar la expresión de MHC en los astrocitos y su participación en la presentación del antígeno a los linfocitosT.
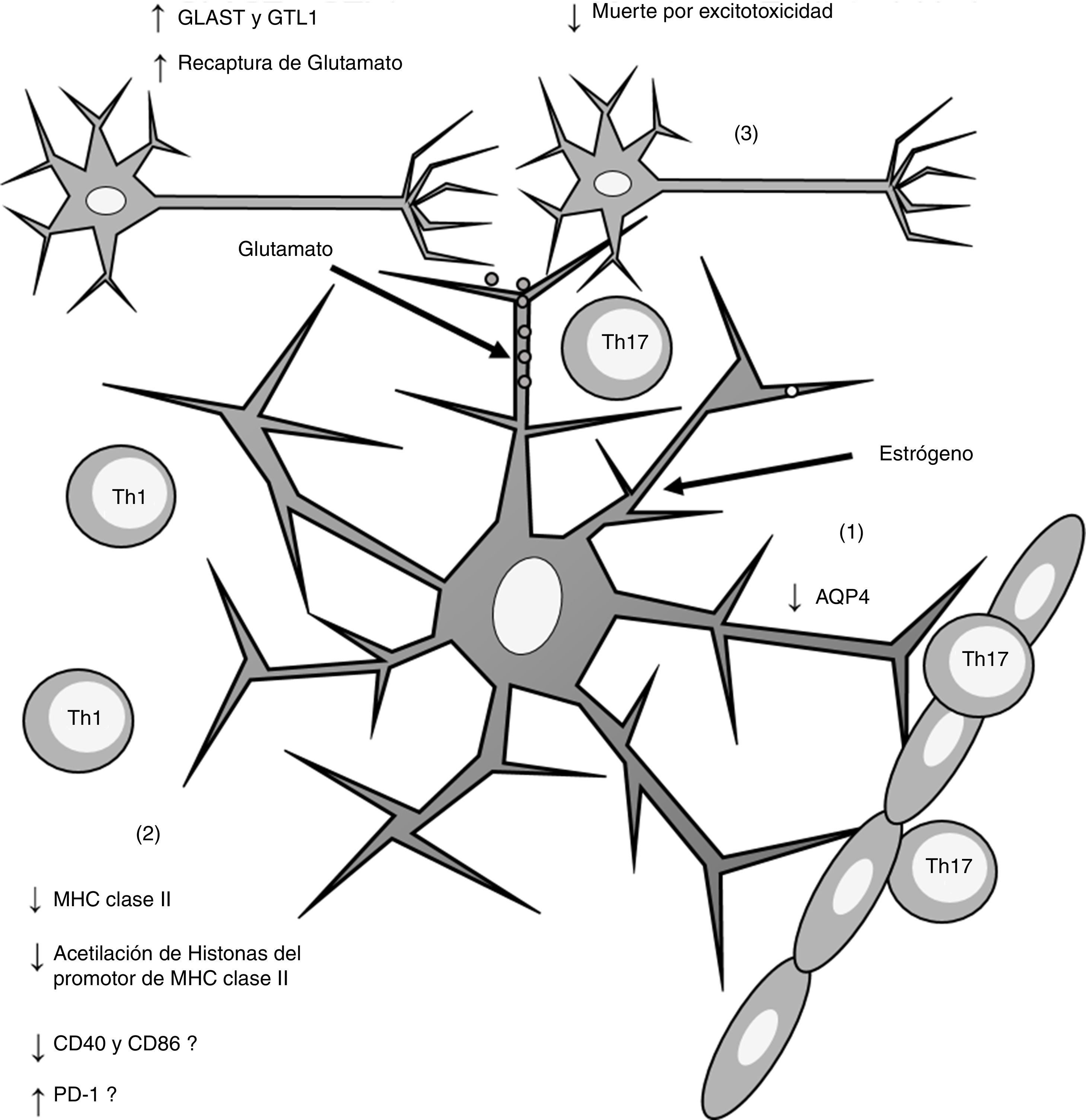
Por otro lado, se ha descrito que el estrógeno induce una respuesta antiinflamatoria en los astrocitos asociada con un efecto neuroprotector que disminuye la severidad de la EAE y la EM66,67 mediante diversos mecanismos. Uno de ellos es la modulación de la actividad de los astrocitos como APC. Adamski et al.68 demostraron que el estrógeno disminuye la expresión del MHC claseii en astrocitos por un mecanismo independiente de CIITA a través de una disminución en la acetilación de histonas localizadas en el promotor del MHC claseii (fig. 2). Además, en microglía, se observó que el estrógeno disminuye la expresión del cluster of differentiation (CD) CD40 y del CD8669, mientras que en APC profesionales (células dendríticas, macrófagos y linfocitosB) el estrógeno incrementa la expresión de la proteína de muerte programada (programmed death-1 [PD-1])70, lo que sugiere que el estrógeno podría también modular la expresión de moléculas coestimuladoras en los astrocitos (fig. 2).
 disminución de la expresión de AQP4; (2) disminución en la expresión de MHC claseii, lo que disminuye la presentación antigénica; (3) incremento en la expresión de los receptores de recaptura de glutamato GLAST y GTL1, lo que incrementa la recaptura de glutamato de la sinapsis neuronal y disminuye la muerte neuronal por excitotoxicidad. AQP4: acuaporina 4; CD40: cluster de diferenciación 40; CD86: cluster de diferenciación 86; MHC: complejo principal de histocompatibilidad; PD-1: proteína de muerte programada-1; Th1: linfocitos Th1; Th17: linfocitos Th17.")
Esquema general de los efectos inducidos por el estrógeno sobre los astrocitos. La administración de estrógeno induce (1) disminución de la expresión de AQP4; (2) disminución en la expresión de MHC claseii, lo que disminuye la presentación antigénica; (3) incremento en la expresión de los receptores de recaptura de glutamato GLAST y GTL1, lo que incrementa la recaptura de glutamato de la sinapsis neuronal y disminuye la muerte neuronal por excitotoxicidad.
AQP4: acuaporina 4; CD40: cluster de diferenciación 40; CD86: cluster de diferenciación 86; MHC: complejo principal de histocompatibilidad; PD-1: proteína de muerte programada-1; Th1: linfocitos Th1; Th17: linfocitos Th17.
Los astrocitos se encuentran en contacto con la lámina basal de la BHE debido a que sus prolongaciones citoplasmáticas rodean las paredes de los vasos sanguíneos71. En este nivel, los astrocitos forman entre ellos uniones comunicantes mediante la expresión de conexina-43 y conexina-3010. Brand-Schieber et al.72 demostraron, en ratones con EAE, que los astrocitos disminuyen la expresión de la conexina-43, lo que sugiere la pérdida de conectividad entre los astrocitos y un aumento de la permeabilidad de la BHE. Por otro lado, los astrocitos expresan acuaporina 4 (aquaporin 4 [AQP4])71, la cual se encuentra sobreexpresada en lesiones activas de EM73 (fig. 1). Recientemente, Soltani et al.74 demostraron en ratas con daño cerebral postraumático que el estrógeno disminuye la expresión de AQP4 (fig. 2), lo que sugiere que el estrógeno podría inducir un efecto protector sobre la BHE en la EAE y la EM.
Además, los astrocitos pueden modular la permeabilidad de la BHE mediante la producción de citocinas. Según las interacciones que tengan los astrocitos con otras células, o los estímulos que reciban de otras citocinas, será la combinación de citocinas que produzcan. En este sentido, la producción de IL-1β, IL-6 y TNF-α por los astrocitos incrementa la permeabilidad de la BHE, mientras que la producción de TGF-β induce el efecto contrario75. Además, la BHE puede transportar citocinas en ambos sentidos76, lo que implica que una producción elevada de citocinas proinflamatorias a nivel sistémico contribuye al estado inflamatorio de la enfermedad subyacente del SNC.
Las células inmunes de la sangre periférica tienen un acceso restringido al interior del SNC. Para ingresar, las células deben migrar por diapédesis a través de la BHE al interior de los espacios perivasculares y después al parénquima del SNC25, por lo que los astrocitos son las primeras células del SNC que son activadas por los linfocitosT, debido a su íntimo contacto con la BHE77. La producción de IL-17 por los linfocitos Th17 y la posterior señalización de IL-17 mediada por Act1 induce la producción de quimiocinas que incrementan el reclutamiento y la diapédesis de células inmunes a través de la BHE77,78. De este modo, la interacción de los linfocitos Th17 con los astrocitos podría ser un evento desencadenante en los procesos inflamatorios de la EM, debido a la activación de los astrocitos y la producción de factores solubles (quimiocinas y citocinas), los cuales provocan un incremento en la permeabilidad de la BHE.
Papel de los astrocitos en la expresión de los perfiles Th1/Th2/Th17Los linfocitos Th1 y Th17 son los linajes de linfocitosT CD4+ responsables de la inflamación en la EM, mientras que los linfocitos Th2 y los Treg limitan la actividad inflamatoria79. Existen diversos estudios en relación con el papel que desempeñan los astrocitos sobre la modulación de los perfiles Th1/Th2/Th17 en el interior del SNC. En este sentido, se ha descrito que los astrocitos producen citocinas de la familia IL-12, como IL-1280,81, IL-23 (ambas subunidades p4080,81 y p1981) e IL-27 (ambas subunidades p28 y EBI3)82. La producción de estas citocinas en la EM, como se mencionará a continuación, parece determinar cuál es el perfil que predomina durante las fluctuaciones de la enfermedad.
Por un lado, la producción de IL-12 e IL-23 favorece la presencia de linfocitos Th1 y Th1783, respectivamente. Debido a que ambas citocinas comparten la subunidad p40, estudios previos han demostrado que IL-23 es esencial para el desarrollo de la EM, ya que la presencia de Th17 inducida por IL-2384, en ausencia de Th1, es suficiente para inducir la EAE en animales85,86. Es por lo anterior que existe una correlación entre la presencia de IL-23 con la severidad de la enfermedad inducida por la producción de IL-1784, la cual provoca la excitotoxicidad y muerte neuronal causadas por la acumulación de glutamato en la sinapsis neuronal65,87 (fig. 1). Por otra parte, cuando se favorece la producción de IL-12, la población predominante serían los linfocitos Th183, por lo que la producción de IFN-γ inhibe la diferenciación a linaje Th1788. La evidencia anterior sugiere que la producción de IL-23 por los astrocitos favorece la inflamación del SNC a través de mecanismos dependientes de Th17 y que la producción de IL-12 promueve parcialmente la inflamación a través de Th1. Sin embargo, una elevada producción de IL-12 sugiere que la prevalencia de la población predominantemente sería Th1, que en junto con la subsecuente producción de IFN-γ participa en la regulación de la actividad de la población Th17.
Respecto a la excitotoxicidad inducida por glutamato y mediada por IL-17, diversos estudios han demostrado que el estrógeno incrementa, en astrocitos, la expresión del receptor transportador de glutamato y aspartato (glutamate aspartate transporter [GLAST])66,89-91 y del transportador de glutamato-1 (glutamate transporter-1 [GLT1])66,90-92. Lo anterior sugiere que el estrógeno ejerce un efecto neuroprotector en los astrocitos a través del incremento en la recaptura del glutamato en la sinapsis neuronal66,90 que puede disminuir la muerte neuronal causada por excitotoxicidad inducida por glutamato (fig. 2).
Por otro lado, IL-27 es producida de forma abundante por los astrocitos y la microglía en pacientes con EM, y su receptor está expresado tanto en linfocitosT como en astrocitos82. IL-27 tiene efectos principalmente antiinflamatorios en la EM, y se ha descrito que limita el desarrollo de la EAE en animales, ya que regula la respuesta de los linfocitos Th1 y Th2 a través de la disminución de IL-2, lo que limita la proliferación de estas poblaciones83. Además, IL-27 disminuye la diferenciación de linfocitos Th1793,94 y puede inducir, junto con IL-6, la producción de IL-10 en linfocitos Th1, Th2 y Th1795. La evidencia anterior sugiere que los astrocitos pueden modular la respuesta inflamatoria de la EM mediante varios mecanismos que incluyen la secreción de IL-27 por los astrocitos y la microglía, lo que provoca un proceso autolimitante debido a la producción de IL-10 por parte de las distintas clonas de linfocitosT.
ConclusionesLos astrocitos desempeñan un papel trascendental en la patogénesis de la EM, debido a que pueden participar directamente en la inflamación del SNC, así como en la limitación de la misma, a través de diversos mecanismos que involucran: a)la expresión de receptores TLR que le permiten actuar como células inmunes; b)la expresión de MHC clasei yii, así como de moléculas coestimuladoras, lo que permite a los astrocitos funcionar como APC profesionales; c)la regulación de la permeabilidad de la BHE, y d)la producción de IL-12, IL-23 e IL-27 que regulan la expresión de los perfiles inflamatorios Th1 y Th17 en el SNC. Además, la evidencia demuestra que el estrógeno induce un efecto neuroprotector sobre los mecanismos en los que participan los astrocitos. Futuros estudios deberán enfocarse en el papel de los astrocitos y la modulación de su actividad en los procesos inflamatorios del SNC con el objetivo de encontrar nuevos blancos terapéuticos para el tratamiento de la EM.
Conflicto de interesesEl autor declara que no se presentó ningún conflicto de intereses en la realización del presente artículo de revisión.
El autor desea agradecer al Instituto Mexicano del Seguro Social por la Beca de Doctorado otorgada; al Dr. Daniel Ortuño-Sahagún y a Karla Itzel Padilla Chavoya por su apoyo en la revisión del presente artículo.

