




The CLTC gene encodes the clathrin heavy chain 1 (CHC1) protein.1,2 This structure enables the formation of lattices in clathrin-coated vesicles by facilitating the intracellular membrane traffic of receptors, endocytosis of certain macromolecules, and stability of the mitotic spindle during the metaphase.3 This protein is expressed in greater abundance in the developing brain.4 Loss-of-function (LoF) mutations of the CLTC gene are associated with autosomal dominant mental retardation-56 (MIM#617854), although they have also been reported in patients with epilepsy and other neurodevelopmental disorders.3–5
We present the case of a girl with a previously unreported de novo mutation of the gene and periventricular heterotopia detected with a brain magnetic resonance imaging (MRI) study.
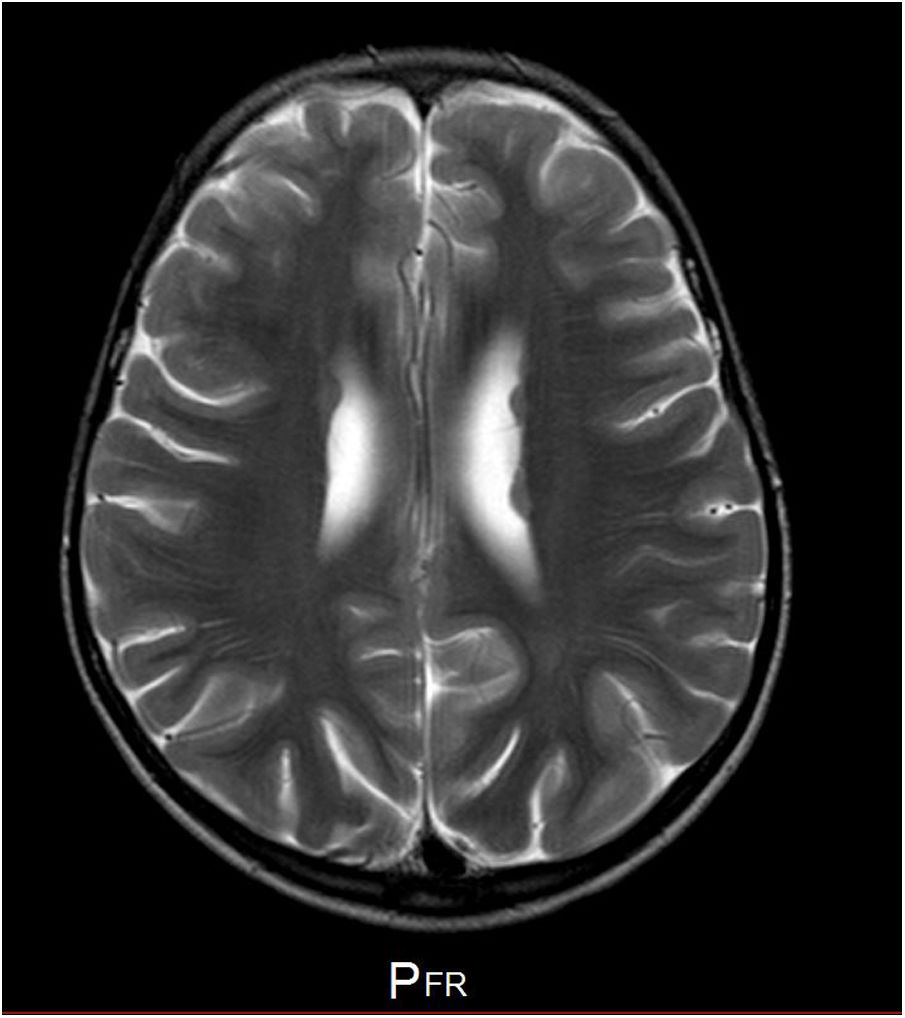
Our patient is a girl with no relevant family history, and personal history of patent ductus arteriosus and bone alterations (spina bifida occulta and mild rib hypoplasia). Head circumference has consistently been in the 10th percentile. Fig. 1 shows the phenotype. At the age of 5, she began to present epileptic seizures (typical absence and generalised tonic-clonic seizures), which were controlled with ethosuximide and valproic acid; seizures returned when the drug was withdrawn when the patient was 7 years old. An EEG performed before treatment revealed 3-Hz spike-and-wave discharges triggered by hyperventilation, which lasted 9seconds. An MRI scan showed periventricular heterotopia (Fig. 2). The patient showed difficulties with attention, language, and reading. Her intelligence quotient according to the Wechsler Intelligence Scale for Children (fifth edition) was 68, with working memory and attention abilities being particularly impaired.


A whole-genome sequencing (WGS) study revealed heterozygous presence of a de novo nonsense mutation in exon 23 of the CLTC gene (hg19; chr 17: 57760140; NM_004859.3; c.3751C>T, p.Arg1251*). The mutation, which was subsequently confirmed by Sanger sequencing, was not listed on any genetic database and had not previously been reported in the literature.
The literature includes nearly 30 cases of de novo LoF variants of the CLTC gene.1 The phenotype is characterised by subtle but consistent dysmorphic features, which can be observed in the patient we present.1 One of these features is bulbous tip of the nose.1 Bone deformities have been described previously.4 The degree of intellectual disability is variable, from borderline intelligence quotient to moderate disability.1,5 Delayed language acquisition and attention deficit/hyperactivity disorder are very frequent.1,5 Gait alterations (ataxic, hypotonic, or spastic) may also present.4,5 Some patients develop parkinsonism in adulthood.4 Corpus callosum hypoplasia is the most frequent structural brain anomaly,1 although microcephalus (10th percentile in our patient), other anomalies (especially of neuronal migration), and epilepsy, as in our patient and other published cases, are also frequent.1,4,5 Absence seizures, as observed in our patient, have previously been described in another patient with a de novo frameshift mutation of the CLTC gene.5 The literature includes reports of adequate seizure control with valproic acid.5 The phenotypic variability observed in patients with LoF variants of this gene is associated with allelic heterogeneity, although the correlation with the type of mutation or the affected segment of clathrin is not well established.1,4
In conclusion, we report a new pathogenic variant of the CLTC gene, to our knowledge never previously reported, in a patient who also presents periventricular heterotopia, contributing to further expansion of the clinical spectrum of mutations of this gene and their association with alterations in neuronal migration.
FundingThe authors have received no funding for this study.
Please cite this article as: Martín Fernández-Mayoralas D, Muñoz Jareño N, Alba Menéndez A, Fernández-Jaén A. Heterotopías periventriculares: ampliación del espectro clínico de las variantes patogénicas del gen de la clatrina 1 (CLTC). Neurología. 2021;36:327–329.
