




El gen CLTC codifica la cadena pesada de la clatrina 1 (CHC1)1,2. Esta estructura permite la formación de las envolturas de las vesículas recubiertas de clatrina, facilitando el tráfico intracelular de receptores, la endocitosis de ciertas macromoléculas y la estabilidad del huso mitótico durante la metafase3. Esta proteína se expresa con mayor abundancia en el cerebro en desarrollo4. Las mutaciones de pérdida de función (loss-of-function [LoF]) del gen CLTC se asocian con un patrón de herencia autosómico dominante al síndrome de discapacidad intelectual tipo 56 (MIM#617854), si bien se ha descrito en pacientes con epilepsia y otras alteraciones del neurodesarrollo3–5.
Exponemos el caso clínico de una niña con una mutación de novo de este gen, no descrita previamente, y heterotopías periventriculares en la resonancia magnética (RM) cerebral.
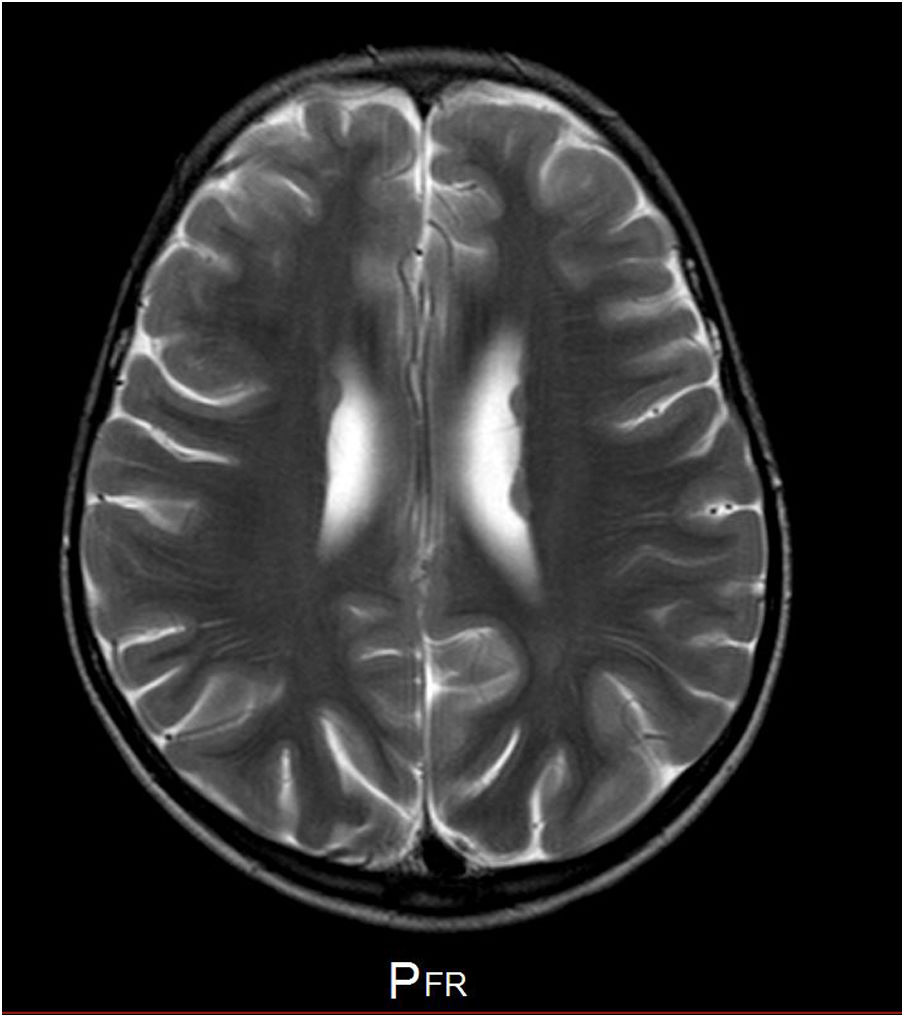
Se trata de una niña sin antecedentes familiares de interés, con antecedentes personales de ductus arterioso persistente y alteraciones óseas consistentes en una espina bífida oculta y una leve hipoplasia costal. El perímetro craneal corresponde de forma constante a un percentil 10. La descripción del fenotipo se presenta en la figura 1. A la edad de 5 años comienza con crisis epilépticas (ausencias típicas y tónico-clónico generalizadas) controladas con etosuximida y ácido valproico (recidivaron tras la retirada del fármaco con 7 años). El EEG previo al tratamiento mostró una punta-onda a 3Hz por segundo, desencadenada con la hiperventilación de 9s de duración. La RM muestra heterotopías periventriculares (fig. 2). La paciente manifestó dificultades en la atención, el lenguaje y la lectura. Presenta un cociente intelectual según la escala de inteligencia de Wechsler para niños-V de 68, con una memoria de trabajo y habilidades atencionales particularmente afectadas.


El estudio por secuenciación completa del genoma en trío (WGS) demostró una mutación de novo, nonsense, en el exón 23, en heterozigosis del gen CLTC (hg19; chr 17: 57760140; NM_004859.3; c.3751C>T, p.Arg1251*). Esta mutación, confirmada posteriormente por Sanger, no ha sido previamente descrita en las bases de datos ni en la bibliografía médica.
Hay cerca de 30 casos publicados con variantes LoFde novo de CLTC1. El fenotipo se caracteriza por rasgos dismórficos sutiles, pero consistentes y ajustados a la de la paciente estudiada en el presente caso clínico1. La punta bulbosa de la nariz es característica1. Las deformidades óseas están descritas previamente4. La discapacidad intelectual es de grado variable, oscila entre el cociente límite y la discapacidad intelectual moderada1,5. El retraso en la adquisición del lenguaje y el TDAH son muy frecuentes1,5. Pueden existir alteraciones de la marcha (atáxica, hipotónica o espástica)4,5. Algunos pacientes han desarrollado parkinsonismo durante la edad adulta4. La hipoplasia del cuerpo calloso es la anomalía cerebral estructural más frecuente1, suele existir microcefalia (centil 10 en el caso mostrado), otras anomalías (especialmente de la migración neuronal) y epilepsia, como en el caso que nos ocupa y otros publicados1,4,5. La epilepsia con ausencias, tal y como padecía la paciente, ha sido descrita previamente en otro caso con una mutación frameshiftde novo del gen CLTC5. El control adecuado con ácido valproico de las crisis también ha sido descrito5. La variabilidad fenotípica observada en individuos con variantes LoF de este gen, se asocia con la heterogeneidad alélica, aunque la correlación con el tipo de mutación o el segmento afectado de la clatrina no está bien establecida1,4.
En conclusión, describimos un caso clínico con una nueva variante patogénica del gen CLTC, siendo el primero hasta nuestro conocimiento, que cursa además con heterotopías periventriculares, lo que contribuye a ampliar aún más el espectro clínico de las mutaciones de este gen y su relación con las alteraciones de la migración neuronal.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.

