



La acetiltransferasa de histonas KAT6B es un componente del complejo proteico de lectores epigenéticos MOZ/MORF. Esta proteína participa tanto en la activación como en la represión transcripcional, habiéndose implicado en el desarrollo cortical cerebral1,2. Al igual que otros genes responsables de la regulación cromatínica, su disfunción genera un trastorno de desarrollo multisistémico1.
Los trastornos conocidos y relacionados con el gen KAT6B incluyen el síndrome genitopatelar (GPS) y el síndrome Say-Barber-Biesecker-Young-Simpson (SBBYSS), también llamado variante Say-Barber-Biesecker del síndrome de Ohdo1,3–8.
Describimos el caso de un niño en el que se identificó una mutación de novo en el gen KAT6B, con un fenotipo compatible con un SBBYSS, sin discapacidad intelectual, pero con un trastorno específico del lenguaje, asociado a problemas atencionales severos.
Se trata de un varón de 8,5 años, hijo único de padres sanos, españoles. Acude a la consulta por problemas en el aprendizaje escolar, dificultades lingüísticas y atencionales marcadas; no mostraba intereses restringidos ni esterotipias, y su interés social era marcado. Su vocabulario era pobre y en su discurso destacaban importantes defectos sintácticos. La historia familiar no aportó datos relevantes. Nacido de un embarazo y parto normales, con peso de 2.970g (pc 10) y talla de 49cm (pc 25) al nacimiento. En su historia personal destaca la cirugía por hipospadias y criptorquidia. En el desarrollo cognitivo resaltaba la presencia de un retraso severo del lenguaje, diciendo escasas palabras con 3 años. El desarrollo motor se situaba en la media, habiendo empezado a caminar con 14 meses. A la edad de 6 años, se realizó una primera evaluación neuropsicológica que reveló en el WPPSI-III un coeficiente intelectual (CI) verbal de 58 (pc 0,3) —subtest «comprensión» con puntuación típica de 1— y manipulativo de 93 (pc 33); no se pudo calcular el CI total por la diferencia entre escalas.
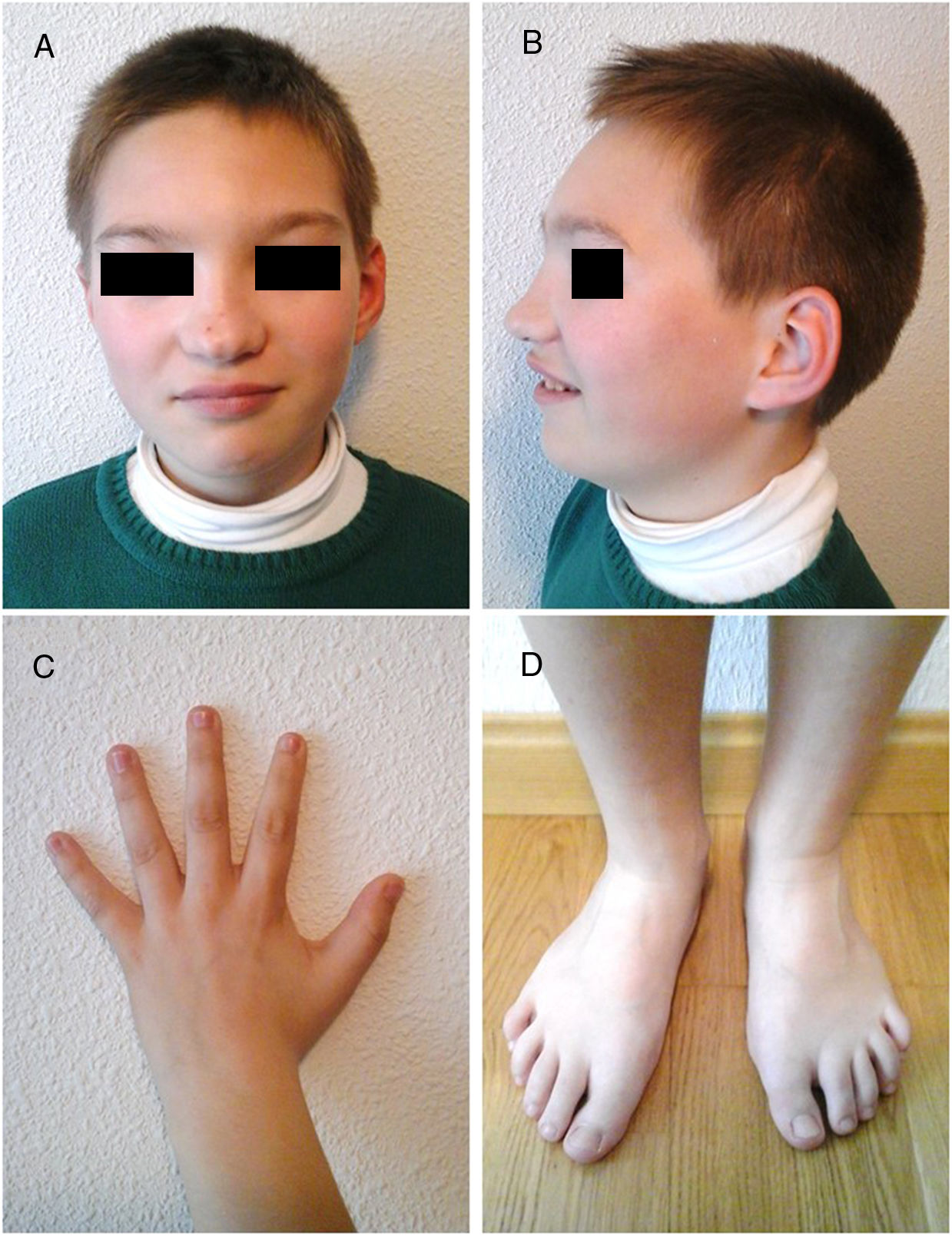
En la exploración física no se objetivó focalidad neurológica, si bien presentaba marcados problemas del lenguaje (pobre vocabulario y comprensión). Tenía un peso de 38kg y una talla de 140cm (pc 97). Asociaba ciertos rasgos dismórficos: blefarofimosis, ptosis palpebral, hipertelorismo, nariz bulbosa, leve retrognatia, limitación para la separación de dedos en ambas manos, leve limitación para la extensión completa de rodilla y dedos largos en ambos pies (figs. 1A-D).
 Rasgos faciales. Blefarofimosis, ptosis, hipertelorismo, nariz bulbosa y leve retrognatia. C) Dorso de mano. Limitación de la abducción del pulgar. D) Dorso de pies. Dedos largos en ambos pies.")
Los estudios genéticos por arrays y la secuenciación de los genes PTEN y NSD1 arrojaron resultados normales. La resonancia magnética cerebral fue normal.
La evaluación neuropsicológica mostró un CI de 108 y 102 en las escalas de Toni-2 y Leiter-R, respectivamente; la evaluación del lenguaje mostró una edad psicolingüística ≤6 años (ITPA, Peabody y BLOC-R). En tareas de ejecución continuada se objetivaron problemas atencionales severos (omisiones y variabilidad del HitRT>pc95).
A los 10 años, se amplió el estudio con análisis trío del exoma completo identificándose una mutación de novo, nonsense, en el exón 16 del gen KAT6B (hg19; chr 10: 76781739; NM_012330.3; c.3122C>A, p.Ser1041*). Esta mutación, confirmada posteriormente por Sanger, no ha sido previamente descrita en las bases de datos ni en la bibliografía médica.
Tras este hallazgo, se realizó evaluación tiroidea y cardiológica, que no revelaron hallazgos significativos.
El fenotipo clínico del caso descrito es similar a los previamente reportados en la literatura. El SBBYSS es un síndrome infrecuente, caracterizado básicamente por blefarofimosis y discapacidad intelectual3–8. Hasta la fecha, todos los casos asociaban graves problemas cognitivos. La asociación con otros rasgos o malformaciones es también frecuente: facies hipomímica, nariz bulbosa, alteraciones ligamentosas o esqueléticas o dedos largos3.
Las mutaciones en el gen KAT6B se han vinculado a los síndromes GPS y SBBYSS. Probablemente ambos sean un continuo sindrómico, atendiendo a la ubicación de la mutación; mutaciones ubicadas en los últimos exones tienden a mostrar un perfil clínico más próximo al SBBYSS5,9.
El caso reportado es importante, al tratarse de un caso sin discapacidad intelectual, y mostrar una mutación no previamente descrita. Sugiere igualmente la presencia de mutaciones en genes previamente vinculados a la discapacidad intelectual y autismo, en otros trastornos del neurodesarrollo10. El clínico debería valorar la conveniencia de estudios genéticos complejos en pacientes con graves trastornos del lenguaje10.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.

