





Primary progressive aphasia (PPA) is a clinical syndrome characterised by a progressive decline in language and speech of neurodegenerative origin. Major breakthroughs made in recent years have lent us a better understanding of this syndrome, which may be the first manifestation of any of a number of neurodegenerative diseases.
DevelopmentWe reviewed the main aspects of PPA epidemiology, clinical manifestations, diagnosis, aetiology and treatment. Most cases manifest sporadically and the typical age of onset is between 50 and 70 years. Three clinically distinct variants have been described: nonfluent or agrammatic PPA, semantic PPA and logopenic PPA. Each of these variants tends to be associated with specific histopathological findings, but clinical diagnostic methods are imperfect predictors of underlying pathology. Anatomical and functional neuroimaging can provide useful biomarkers. Several treatments have been proposed, and while no clear benefits have been demonstrated, acetylcholinesterase inhibitors may be useful, especially in the logopenic variant.
ConclusionsPPA is an emerging syndrome which may be more prevalent than we might expect. It was previously listed as part of the frontotemporal dementia spectrum, and it is also related to Alzheimer disease. Clinical diagnosis, complemented by a biomarker evaluation, may predict the underlying pathology, which in turn will improve treatment possibilities.
La APP es un síndrome clínico caracterizado por un deterioro progresivo del lenguaje de etiología neurodegenerativa. En los últimos años se han realizado importantes avances que han contribuido a un mayor conocimiento de esta entidad, que puede ser el modo de presentación de diferentes enfermedades neurodegenerativas.
DesarrolloSe revisan los principales aspectos epidemiológicos, clínicos, diagnósticos, etiológicos y terapéuticos de la APP. La mayoría de los casos son esporádicos, iniciándose en torno a los 50-70 años. Se han descrito 3 subtipos clínicos principales: APP-no fluente o agramatical; APP-semántica; y APP-logopénica. Cada subtipo se ha asociado de forma preferencial a una anatomía patológica concreta, aunque la capacidad predictiva del diagnóstico clínico no es completa. Entre los biomarcadores disponibles, destacan la neuroimagen anatómica y funcional. Se han ensayado diferentes tratamientos, sin demostrarse un beneficio claro. No obstante, los inhibidores de acetilcolinesterásica pueden estar indicados, especialmente en la variante logopénica.
ConclusionesLa APP es un síndrome emergente, con una prevalencia probablemente mayor de la esperada. Considerada previamente como parte del espectro de la demencia frontotemporal, también está relacionada con la enfermedad de Alzheimer. El diagnóstico clínico, completado con el uso de biomarcadores, puede predecir la anatomía patológica subyacente, lo que a su vez supondrá mayores oportunidades en el tratamiento.
Progressive primary aphasia (PPA) is a clinical syndrome of neurodegenerative origin characterised by an insidious deterioration of language ability. For PPA to be diagnosed, language alteration must be present as the main clinical manifestation during at least the first 2 years of the disease. No other cognitive domains are altered (memory, visuospatial abilities) and any limitations in activities of daily life are related to language dysfunction and not to other functions. This heterogeneous syndrome, which was recently described by Mesulam in the literature, may be the form of presentation of any of a number of different neurodegenerative diseases. In recent years, special emphasis has been placed on categorising PPA in different clinical forms and describing biomarkers. These data could contribute to predicting the condition's anatomical pathology. This article aims to review the main advances in our understanding of PPA in recent years. These advances have led to substantial changes in the clinical approach to the syndrome in addition to raising interesting perspectives needing further study.
Epidemiology and risk factorsThe onset of PPA tends to occur between the ages of 50 and 70 years.1 There are no clear distinctions between cases in males and females; different studies point to slightly higher frequencies in men2 or in women.3 While its frequency in the general population is unknown, it can be extrapolated from available data on patients with frontotemporal dementia (FTD). The prevalence of FTD is calculated as approximately 5 cases per million inhabitants4 with between 1 and 15 cases per 100000 inhabitants in individuals younger than 65 years.5–7 PPA accounts for 20% to 40% of these cases. A number of epidemiological studies on dementia in Spain8,9 have estimated FTD prevalence at 0.2% to 0.3% among individuals older than 65 years.10,11 Other studies have suggested a higher prevalence of PPA in patients with learning disorders such as dyslexia. This trend might be explained by these patients or their families having more susceptible language-related neural networks.12 A higher rate of vasectomies has also been measured in this population, which might suggest an autoimmune basis.13 However, the above associations have not been found by other studies.
The vast majority of PPA cases are sporadic.14,15 Some patients belong to families with mutations on chromosome 17, either in the gene coding for microtubule-associated protein tau (MAPT) or the gene coding for progranulin.16,17 Cases of tau pathology, which typically arise in the context of FTD with behaviour disorders,18 are associated with MAPT mutations that are often associated with a gain in toxic function. On the other hand, progranulin mutations are related to forms of frontotemporal lobar degeneration with ubiquinated inclusions (FTLD-U). Progranulin mutations typically present as non-fluent or logopenic aphasia,16,18,19 whereas MAPT mutations mainly present as non-fluent or semantic aphasia. Behavioural changes are commonly associated in the latter case.18,20 Nevertheless, the different mutations may manifest with varying phenotypes, and finding distinct syndromes is typical (progressive aphasia, behavioural variant FTD) among members of the same family.16 Other researchers have proposed that progranulin mutations could give rise to a distinctive language disorder that would not match any of the common subtypes.19
ApoE genotype has not been shown to have any predictive value apart from providing a clinical description for diagnosing Alzheimer disease (AD) in PPA in the study with the largest sample size to date.21,22 Although the e4 allele is more frequently present in the logopenic variant than in other clinical forms of PPA, its prevalence overall is lower than in typical AD.22,23 In turn, the H1/H1 polymorphism of MAPT is more commonly found in sporadic PPA.24 It has been proposed as a risk factor for corticobasal degeneration and progressive supranuclear paralysis, and to a lesser extent, for FTD.25–27 Heterozygosity at codon 129 of the prion protein may also entail greater risk.28
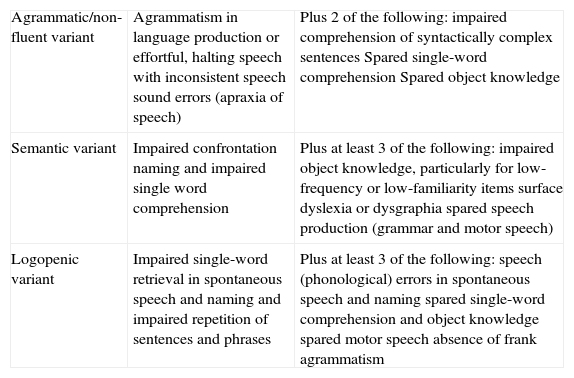
Clinical typesThree main clinical subtypes have been described for PPA, each one associated with a certain pattern of anatomical and functional radiology images.3,29 These subtypes are non-fluent/agrammatic variant (navPPA); semantic variant (svPPA); and logopenic/phonologic variant (lvPPA). The first 2 are included among types of FTD. However, a minority of patients cannot be included in any of these groups (‘non-classifiable PPA’), whether because they present isolated linguistic symptoms or because they show characteristics belonging to more than one group.30 A consensus statement on criteria for diagnosing and classifying PPA according to its 3 main variants was recently published (Table 1).30 While this consensus statement is still pending definitive validation, and not all authors agree, it aims to eliminate the disparities that existed previously between different centres and studies, and which made extrapolating results a difficult task.
Clinical diagnostic criteria for the 3 PPA variants.
| Agrammatic/non-fluent variant | Agrammatism in language production or effortful, halting speech with inconsistent speech sound errors (apraxia of speech) | Plus 2 of the following: impaired comprehension of syntactically complex sentences Spared single-word comprehension Spared object knowledge |
| Semantic variant | Impaired confrontation naming and impaired single word comprehension | Plus at least 3 of the following: impaired object knowledge, particularly for low-frequency or low-familiarity items surface dyslexia or dysgraphia spared speech production (grammar and motor speech) |
| Logopenic variant | Impaired single-word retrieval in spontaneous speech and naming and impaired repetition of sentences and phrases | Plus at least 3 of the following: speech (phonological) errors in spontaneous speech and naming spared single-word comprehension and object knowledge spared motor speech absence of frank agrammatism |
The agrammatic variant is characterised by hypofluent and laboured language production containing grammatical errors, phonological paraphasias, difficulty understanding complex grammatical structures, and occasionally, apraxia of speech. Acalculia with ideomotor and orofacial apraxia may also be present; all are mild, at least in initial stages. Prosody may also be altered.
The semantic variant of PPA is caused by progressive loss of semantic understanding.31 This manifests clinically as fluent, grammatically correct language with impaired confrontational naming, diminished comprehension of simple words, and poor object recognition. Speech becomes progressively devoid of meaning and employs circumlocutions and semantic paraphasia. However, the abilities to understand and repeat phrases are spared. In addition, superficial dyslexia, associative visual agnosia, and prosopagnosia may be present.32 Mesulam considers it convenient to distinguish between patients with a strictly linguistic disorder (semantic PPA) and those whose condition is associated with visual agnosia or other recognition-related deficiencies (semantic dementia) such as tactile or gustatory agnosia.33,34 This opinion is not shared by other authors, who believe that svPPA and semantic dementia refer to the same entity with identical anatomical pathology features, although probably at different stages of progression.35
The logopenic variant is characterised by markedly anomic language containing frequent pauses. The overall result is slow speech and decreased fluency with frequent pauses to search for words. As a result, it may be difficult to distinguish between this variant and the agrammatic variant.23,36 Articulation, prosody, and grammar remain intact,30,37 and so does comprehension of basic words. In contrast, sentence repetition ability is affected; this is one of the 2 main characteristics of lvPPA, along with difficulty finding words. For these reasons, the variant resembles conduction aphasia. Unlike in svPPA, patients with logopenic aphasia are typically able to point to an object named by an examiner, and can describe (or at least mime) how to use it. The disorder seems to be secondary to damage in the phonological circuit of working memory; in such cases, the meaning of words remains intact.
Although the clinical variants are defined according to aphasia semiology, there are other associated symptoms that can help provide a diagnosis. For example, behavioural changes similar to those in behavioural variant FTD (bvFTD) are frequently found with the semantic variant, but not with the agrammatic or logopenic variants. Particularly common changes include loss of inhibition, abnormal motor behaviour, and eating disorders.38 Extrapyramidal signs in the initial neurological examination are more common in the agrammatic and logopenic variants; they rarely appear in the semantic variant.31,39 Specifically, bradykinesia (especially hypomimia) and rigidity (appendicular and bilateral in particular) are more common in the agrammatic variant, while gait alterations39 (less specific than those in extrapyramidal disorders) appear in the logopenic variant. Therefore, behavioural changes and mild parkinsonism may be useful for distinguishing between clinical variants in difficult cases, even if such signs are not listed as diagnostic criteria.
Progression and prognosisAs in Alzheimer disease, researchers of APP refer to a preclinical, prodromal phase that may be called ‘mild aphasic cognitive impairment’. However, the usefulness of this distinction is not clear. Memory disorders are common in normal ageing, and distinguishing between normal and pathological changes is therefore crucial. In contrast, changes in language ability are always pathological and may not be attributed to ageing alone.15
Age at onset tends to fall between 50 and 70 years,40 although the age range may be far wider.7 Once it is established, the aphasic disorder tends to progress, developing into mutism in many cases.40 Loss of independence occurs at a later stage than in other types of dementia (at 7 years in 50% of all patients, according to LeRhun et al.). In contrast, survival is similar, ranging from about 7 to 10 years,40 although recent studies suggest that the course of APP may be more gradual.41 In some patients, language deficiency remains the predominant clinical manifestation, or even the only one. Others may present additional deficiencies including cognitive, behavioural, or extrapyramidal deficits. We do not know how often patients with PPA develop generalised dementia or additional deficiencies (named ‘PPA plus’14,15) in the course of this disease. In early stages, patients may exhibit acalculia and ideomotor/visuoconstructive apraxia, but for a diagnosis of PPA, any such deficiencies must be mild and they cannot be the cause of the patient's functional limitations. In a longitudinal follow-up study by Kertesz et al.,42 54% of the sample developed a second or third syndrome other than PPA (FTD, PSP, or CBD) during monitoring.
Disease progression may be monitored clinically, although using scales typically employed for measuring AD may be difficult, considering how much language affects other cognitive domains.43 Addenbrooke's Cognitive Examination may be useful for diagnosis and for following disease progression.44 Additionally, a specific Progressive Aphasia Severity Scale (PASS)45 has been designed to evaluate 10 language domains by means of a clinical interview with both patient and caregiver. The score has been correlated with the degree of selective atrophy.46
Anatomical pathologyThe time requirement of 2 years during which aphasia is the dominant feature is applied in order to exclude patients with Creutzfeldt-Jakob disease and typical forms of AD that initially affect language. While useful, this requirement has not been clearly validated by longitudinal studies,47 and it does not manage to simplify anatomical pathology data, which is heterogeneous in PPA. Underlying pathologies may be classified in 3 large groups: tau diseases of the frontotemporal lobar degeneration spectrum, including Pick disease, PSP, CBD, and other less frequent entities including multisystem tauopathy or argyrophilic grain disease; tau-negative, ubiquitin-positive frontotemporal lobar degeneration mainly related to TDP-43 (FTLD-TDP43); and AD. Less frequently, we find dementia with no distinctive histopathological signs, or Lewy body dementia.
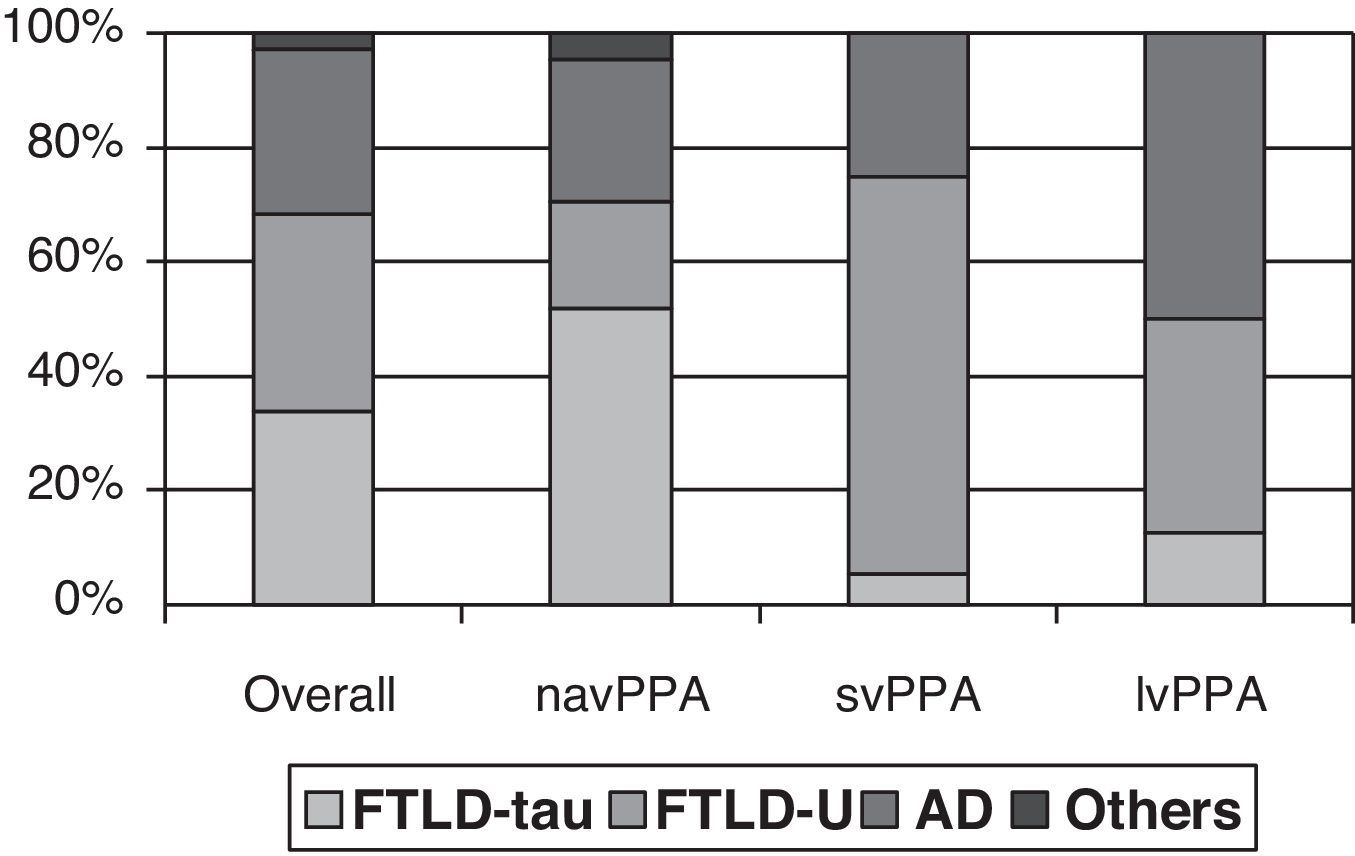
Clinical-pathological correlation studies have shown that each of the clinical forms exhibits a preferential association with an anatomical pathology profile. In the review by Grossman,7 which included 145 cases from 7 different series, non-fluent progressive aphasia was shown to be more associated with tau-positive FTLD, semantic dementia with FTLD-U, and logopenic aphasia with AD. Although the clinical form can only be associated with a specific disease in 50% to 70% of all cases, distinguishing between clinical subtypes increases our ability to predict the underlying disease (Fig. 1). However, most studies focus on one or two PPA subtypes, and diagnostic criteria for different clinical forms are not the same. This yields contradictory results in some cases.21,31,42,47–51 In fact, some authors support classifying patients within the agrammatical PAA category to distinguish between cases of apraxia or true dysarthria (associated with tauopathy) and cases of agrammatic aphasia (associated with FTLD-ubiquitin).50,52,53

Anatomical pathology study of PPA. Taken from data by Grossman.7
Some have attempted to link specific clinical, neuropsychological, and complementary findings to underlying pathologies, as shown in Table 2. For example, apraxia of speech or extrapyramidal signs may be associated with FTLD-tau, while early onset episodic memory loss seems to be linked to AD. The appearance of motor neuron signs is probably the most specific finding, and it indicates FTLD-U. Demographic data, such as age at onset and survival times, have also been evaluated. Studies have revealed a tendency towards earlier onset in cases with tau-negative FTLD42,54 compared to tau-positive FTLD and AD. This finding, however, has not been highlighted by other studies, and it may have to do with the subgroup of patients with motor neuron diseases. Survival seems not to differ between these groups. Although anatomical pathology predictions based on individual data may lead to errors,21 contemplating clinical, neuropsychological, and neuroimaging findings (anatomical or functional) as a whole may increase the probability of finding the correct answer.56 However, studies validating this approach in PPA are still lacking.
Manifestations and findings that may suggest a specific anatomical pathology in PPA.
| Manifestations or findings | Anatomical pathology suggestive of: |
| Clinical signs | |
| Apraxia of speech associated with aphasia | Tauopathy (especially CBD)49 |
| Dominant or isolated apraxia of speech | Tauopathy (especially PSP)49 |
| Supranuclear paralysis of vertical gaze with slow vertical saccades | PSP49 |
| Early behavioural changes in the semantic variant | FTD (AD therefore unlikely)31,54 |
| Signs of a motor neuron disease | FTLD-ubiquitin31 |
| Extrapyramidal signs | Tauopathy39,55,100 |
| Neuropsychological | |
| Early memory loss, visual memory impairment | AD42,62 |
| Logopenic PPA with a low score on memory tests | Alzheimer disease21,42 |
| Executive dysfunction (visual construction, reverse digit span) | Tauopathy (compared to AD and TDP-43)62,100,101 |
| Pronounced deficit in phonemic verbal fluency (Controlled Oral Word Association Test) | More in FTLD-U than in AD62 |
| Imaging | |
| Atrophy and hypoperfusion or temporoparietal hypometabolism (especially when bilateral) in the hypofluent variant. | AD68 |
| Left temporoparietal and left frontal lobe atrophy compared to anterior temporal lobe atrophy | AD (rather than FTLD-U)62 |
| Left medial frontal and left anterior temporal lobe atrophy compared to parietal | FTLD-U (rather than AD)62 |
One controversial topic is the role AD plays in PPA57: although the disease is identified in many autopsies, some scholars doubt that it would cause PPA.21,58,59 While the pattern of atrophy is predominant in language areas, the distribution of neurofibrillary tangles and neuritic plaques is no different from that in patients with AD who present typical amnestic syndrome. In addition, no asymmetry is found between the brain hemispheres. In summary, these cases do not display the numerous typical AD lesions in the areas that would explain the patient's clinical presentation with an aphasic disorder. However, not all studies coincide on this point.47,60 In any case, this finding raises the possibility that AD is not the true cause of these patients’ symptoms. Two neurodegenerative diseases may merely coexist in the same patient as a possible result of common causes or shared aetiological or pathogenic factors. According to this hypothesis, AD may mask the true underlying anatomical pathology features in PPA. With this in mind, the finding of argyrophilic thorny astrocyte clusters in patients with both PPA and AD, but not in patients with typical AD,58 may support the hypothesis of parallel neurodegenerative processes. It must be stated that this finding has not been highlighted by any subsequent studies.59 With a similar objective in mind, researchers have also investigated TDP-43 inclusions based on a suspicion of FTD anatomical pathology masked by AD. What they found, however, was a low percentage of cases with both PPA and AD.59 If, on the other hand, symptoms stem from focal onset of AE, as many authors have professed,51,57 we do not know why it adopts this pattern at onset, or if the syndrome's response to cholinesterase inhibitors will resemble that in typical AD, to name just a few unanswered questions.
Anatomical and functional neuroimagingMagnetic resonance imaging has been used in different studies to determine cortical thickness in order to describe atrophy patterns associated with each of the variants. Generally speaking, left frontal inferior and insular atrophy is detected in agrammatical PPA; predominantly left-sided bilateral anterior temporal atrophy in the semantic variant; and left temporo-parietal atrophy in the logopenic variant (Table 3).3 As the disease progresses, atrophy extends to other zones while following a distinct pattern depending on its clinical form.61 Certain atrophy patterns may furthermore predict the underlying anatomical pathology features,62 or the clinical course of the disease.63 With this in mind, studies using tractography with diffusion tensor imaging64–67 have been performed to observe how different language pathways are affected in each variant. In the non-fluent form, researchers observed impairment of the dorsal pathways (superior longitudinal fasciculus), while in logopenic aphasia, the temporoparietal component of the dorsal pathway is the most damaged. These studies clearly show that damage to the white matter and to different neural networks involved in language is present in PPA.64,66 As such, the studies offer useful information about how pathways are involved in language function. It is possible that certain pathways could be associated with specific diseases, considering that studies have shown that some linguistic symptoms are able to predict these diseases.49
Atrophy pattern on MRI.
| Agrammatic PPA | Left inferior frontal lobe and left insular cortexIf mutism is present: left pars opercularis and left basal ganglia. |
| Semantic PPA | Anterior temporal lobe, bilateral but predominantly left-sided |
| Logopenic PPA | Posterior temporal lobe and left inferior parietal lobe |
| Apraxia of speech | Additional left premotor and motor areas |
Studies carried out using positron emission tomography (PET scans), some of which examine the disease which concerns us,68 have shown that focal uptake is present in all progressive forms of aphasia, regardless of the underlying anatomical pathology traits.69–71 Hypometabolism is prerolandic in non-fluent variants and postrolandic in fluent variants.72 There are also different patterns associated with the 3 main clinical variants: frontal lobe hypometabolism (agrammatic PPA), anterior temporal lobe (semantic PPA), and temporoparietal lobe (logopenic PPA). All patterns are predominantly left-sided.71 Temporoparietal hypometabolism, especially when present bilaterally, is suggestive of AD anatomical pathology.68 PET scans with Pittsburgh compound also show that binding is more frequently increased in the logopenic variant, which would be congruent with its more frequent association with AD.71 Studies that have been carried out seem to show that PET has a higher sensitivity than SPECT and anatomical MRI,73 while displaying a focal pattern associated with each PPA variant.71,72 Furthermore, some of these patterns may be able to predict underlying anatomical pathology.68 However, sample size is small in all of these studies, and each set of conclusions will have to be validated.
Biomarkers in cerebrospinal fluidBiomarkers in the CSF have been widely studied in Alzheimer disease, both for confirming the diagnosis and for identifying higher-risk patients during prodromal phases.74,75 Others emphasise their use as a means of evaluating the efficacy of disease-modifying drugs.76 In studies performed to date, the pattern of increased tau protein (total and phosphorylated) and decreased A-Beta-1-42 is a sensitive and specific tool for diagnosing AD. These biomarkers may also be used for diagnosing atypical forms of AD onset, including progressive aphasia. This being the case, they can help us distinguish between AD and other forms of aphasia with a different underlying pathology.77 FTD is associated with a pattern opposite to that found in AD: tau protein levels are low and A-beta-1-42 is normal or elevated.78 The tau protein/A-beta-1-42 ratio may be particularly sensitive and specific for distinguishing between AD and FTD.79,80 Therefore, CSF biomarkers may be used in diagnosing the underlying disease in cases of PPA. Nevertheless, studies have never been carried out in large samples of patients with PPA. For this reason, there is no evidence that these biomarkers would be useful for identifying patients with anatomical pathology signs of AD within this patient subgroup.81–83 In addition, there are no specific markers of frontotemporal lobar degeneration and its various forms (tauopathies, ubiquitin diseases) that would allow us to perform in vivo diagnosis, or at the very least, increase the predictive capacity of clinical examination and other techniques mentioned previously. Some proposals have included chromogranin B, cystatin C, and IL-17.84 All of these tools will be useful as new treatments modifying amyloid beta and tau pathways emerge.
TreatmentsTreatment is based on 3 fundamental pillars: symptomatic drug treatment and/or disease-modifying drugs; language rehabilitation and strategies for improving communication; and support for patients and caregivers.85
At this time, no large-scale clinical trials are underway, and the existing studies, which have small sample sizes and include data from clinical trials in patients with frontotemporal lobe dementia, have not shown any drug treatments to be effective. Pilot studies have tested bromocriptine86, galantamine,87 rivastigmine,88 selegiline,89 and memantine.90,91 Only galantamine seems to demonstrate a (non-significant) tendency towards stabilising aphasia. Other suggested treatments have included corticosteroids,92 but these trials were limited to single cases.93 Although there are no studies of cholinesterase inhibitors in the logopenic variant, it seems logical to try the treatment in suspected cases of AD. On the other hand, considering that many patients are conscious of their illness for years, it is a good idea to screen for depression and treat it if necessary.94
Studies evaluating speech therapy are also lacking, even if the treatment is thought to be useful. Clinical improvements have been demonstrated in individual cases, as shown by activation of preserved cortical areas during therapy, with favourable results for all 3 clinical variants.95–99 Some patients may also be able to learn alternative forms of communication.33
ConclusionsPPA is a clinical syndrome with 3 main semiological variants that correspond to specific anatomical areas. Diagnosis of the entity is becoming increasingly frequent as our understanding grows; in addition, it is clinically useful to the study of neurodegenerative diseases. Furthermore, a significant number of cases of PPA may be erroneously classified as AD, given that some of the most widely used neuropsychological tests evaluate verbal memory (and not other types, such as visual memory), and doctors may rely on the symptom of being unable to find words as a sign of memory and/or language loss. In addition, there may be patients with typical cases of AD who display marked language loss from onset, or patients with PPA who experience significant memory loss in early stages. These forms may overlap, and they are probably more closely linked to AD than to the frontotemporal lobe degeneration spectrum.
Likewise, PPA is a good example of the possibilities and limitations of syndromic diagnosis, since improved clinical descriptions have enabled identification of 3 well-defined variants. Each of these variants is associated with a typical, although not completely defined, anatomical pathology profile. For the above reasons, we must perfect biomarker techniques (anatomical and functional imaging, CSF protein studies) while continuing to expand the clinical and neuropsychological descriptions of each variant. This will deliver a working approximation of an anatomical pathology diagnosis during the patient's lifetime. In fact, in recent years, the improvement in the area of diagnostics has been due to perfecting semiological descriptions rather than to developing complementary analytical or imaging techniques. In any case, approaches must permit correct use of disease-modifying treatments, where available, which will have a positive effect on these patients.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Matías-Guiu JA, García-Ramos R. Afasia progresiva primaria: del síndrome a la enfermedad. Neurología. 2013;28:366–74.
