






Type 1 neurofibromatosis is the most common neurocutaneous syndrome. Most published case series study the paediatric population.
Material and methodsCross-sectional study of cases of type 1 neurofibromatosis from neurology departments that were recorded in a database. We analysed the different clinical variables providing the diagnosis as well as demographic and neuroradiological variables.
ResultsWe found a total of 31 patients with type 1 neurofibromatosis. The mean age was 28.9 years and 58.4% were women. Subjects with unidentified bright objects (UBOs) were younger than those without them (22.45±8.22 years vs. 32.5±10.64; P=.011). In contrast, subjects with neurofibromas were older than those without them (30.56±10.68 years vs. 18.25±4.34; P=.032). No sex differences were found in the presentation of clinical or radiological variables. Seven patients (22.6%) had tumours; 3 were optic pathway gliomas (1 bilateral), 3 were plexiform neurofibromas, and 1 was a pilocytic astrocytoma in the brainstem.
ConclusionsPatients with type 1 neurofibromatosis presented both peripheral neurofibromas and tumorous lesions of the central nervous system. Subjects with neurofibromas were older than those who did not present them, while subjects with UBOs were younger than those without such lesions.
La neurofibromatosis tipo 1 es el trastorno neurocutáneo más frecuente. La mayoría de las series de casos publicadas son sobre la población pediátrica.
Material y métodosEstudio transversal de los casos de neurofibromatosis tipo 1 en las consultas de neurología recogidos en una base de datos. Se han analizado las diferentes variables clínicas que conforman el diagnóstico, así como las variables demográficas y neurorradiológicas.
ResultadosSe han encontrado un total de 31 pacientes con neurofibromatosis tipo 1. La edad media ha sido de 28,9 años y el 58,4% son mujeres. Los sujetos con lesiones tipo Unidentified bright objects (UBO) son más jóvenes que los que no las presentan (22,45±8,22 años vs. 32,5±10,64; p=0,011), por el contrario, los sujetos con neurofibromas son mayores que los que no los tienen (30,56±10,68 años vs. 18,25±4,34; p=0,032). No hay diferencias de sexo en la presentación de las variables clínicas ni radiológicas. Siete pacientes presentaron tumores (22,6%), 3 fueron gliomas del tracto óptico (uno de ellos bilateral), 3 neurofibromas plexiformes y un astrocitoma pilocítico del troncoencéfalo.
ConclusionesLos pacientes con neurofibromatosis tipo 1 no solo presentan lesiones tumorales a nivel periférico en forma de neurofibromas, sino también a nivel del sistema nervioso central. La edad de los sujetos que tienen neurofibromas es mayor que la que no los presentan, sin embargo, los que presentan UBO son más jóvenes que los que no poseen estas lesiones.
Type 1 neurofibromatosis (NF1) was first described by von Recklinghausen in 1882. NF1 is the most common neurocutaneous syndrome and its approximate incidence is 1 per 3000 to 3500 live births.1 It affects both sexes equally. This autosomal dominant inherited disorder has high penetrance and markedly variable clinical expression. About 30% to 50% of the cases are due to de novo mutations. In 1990, the gene responsible for this disease was detected in the long arm of chromosome 17. The protein produced by this gene, neurofibromin, was also identified.2
Diagnosis of NF1 is based on the presence of at least 2 of the criteria established by the National Institute of Health Consensus Development Conference in 1988 (Table 1). However, it is currently diagnosed by identifying the gene mutation of NF1, which is present in 95% of all cases. Most NF1 manifestations are strongly age-dependent; neurofibromas do not usually appear before adolescence and their frequency increases with age. Café-au-lait spots, in contrast, are usually present by the age of 5, but their number begins to decrease at about the age of 50.3 Some studies reveal that NF1 patients present a shorter life expectancy.4
Diagnostic criteria for type 1 neurofibromatosis. Two or more of the following criteria are required.
| 1. Six or more café-au-lait spots with the following diameter: |
| ≥5mm before puberty |
| ≥15mm after puberty |
| 2. Two or more neurofibromas of any type or one plexiform neurofibroma |
| 3. Axillary or inguinal freckling (Cowden syndrome) |
| 4. Optic pathway glioma |
| 5. Two or more Lisch nodules (benign hamartomas of the iris) |
| 6. Typical bone lesions: |
| Sphenoid dysplasia |
| Dysplasia or thinning of long bone cortex (pseudarthrosis) |
| 7. First-degree relatives with NF1 |
In addition, T2-weighted MRI signal changes known as unidentified bright objects (UBOs) have been described in 43% to 93% of children with NF1.5 UBOs are uncommon in NF1 patients older than 20. They are usually located in the globus pallidus, thalamus, hippocampus, and brainstem. Although most of these lesions may remain stable or even disappear, some may transform into gliomas.6
Given that most series examining NF1 cases include paediatric patients, our aim is to ascertain the clinical and neuroradiological characteristics of a series of adults with NF1.
Materials and methodsThis cross-sectional study of patients diagnosed with NF1 and treated in neurology consults. Data from patients’ routine clinical assessments were kept in a database. NF1 was diagnosed according to the criteria of the National Institute of Health Consensus Conference. Patients monitored in our consults were aged 14 years and older.
We recorded the following demographic and clinical data: sex, age, family history, referring department, and the presence of café-au-lait spots, neurofibromas, axillary or inguinal freckling, Lisch nodules, skeletal changes, or neurological symptoms. Similarly, patients’ neuroimaging tests were checked in order to confirm the presence of UBOs,5 optic pathway gliomas, cerebral astrocytoma, stenosis of the aqueduct of Sylvius, and vascular lesions. In patients with lesions, we monitored lesion changes using serial neuroimaging tests.
Quantitative variables were expressed as means and standard deviations, whereas qualitative variables were expressed as percentages. Means and cross tabulations were compared using the chi-square test to determine whether the patients’ clinical and neuroradiological characteristics reflected age or sex differences. We used SPSS software® v. 15.0. The level of statistical significance was set at P<.05
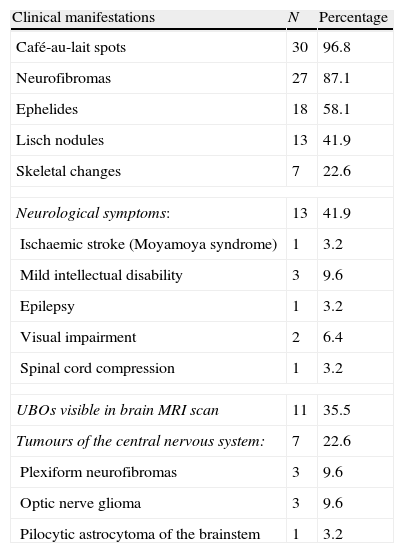
ResultsWe assessed a total of 31 patients with NF1. The mean age was 28.97 (±10.8); there were 17 women (54.8%). We only performed a genetic study of 4 patients (12.9%). Clinical and neuroradiological characteristics of NF1 patients are listed in Table 2. Family history was recorded in 19 subjects (61.3%) The vast majority of patients presented café-au-lait spots (96.8%). Some patients had neurofibromas (87.1%), ephelides (58.1%), and Lisch nodules (41.9%). Seven subjects experienced skeletal changes (22.6%); 5 had scoliosis, 1 had sphenoid dysplasia, and 1 had macrocrania.
Clinical manifestations of NF1 patients (n=31).
| Clinical manifestations | N | Percentage |
| Café-au-lait spots | 30 | 96.8 |
| Neurofibromas | 27 | 87.1 |
| Ephelides | 18 | 58.1 |
| Lisch nodules | 13 | 41.9 |
| Skeletal changes | 7 | 22.6 |
| Neurological symptoms: | 13 | 41.9 |
| Ischaemic stroke (Moyamoya syndrome) | 1 | 3.2 |
| Mild intellectual disability | 3 | 9.6 |
| Epilepsy | 1 | 3.2 |
| Visual impairment | 2 | 6.4 |
| Spinal cord compression | 1 | 3.2 |
| UBOs visible in brain MRI scan | 11 | 35.5 |
| Tumours of the central nervous system: | 7 | 22.6 |
| Plexiform neurofibromas | 3 | 9.6 |
| Optic nerve glioma | 3 | 9.6 |
| Pilocytic astrocytoma of the brainstem | 1 | 3.2 |
Thirteen patients (41.9%) presented clinical neurological manifestations, including ischaemic stroke at the age of 43 secondary to Moyamoya syndrome (1 patient), childhood onset migraine (5), mild intellectual disability (3), complex partial epilepsy with secondary generalisation (1), visual impairment with decreased visual acuity due to an optic nerve tumour (2), and Brown-Séquard syndrome due to dorsal spinal cord compression caused by the neurofibroma (1). All patients were studied with brain MRI. That procedure was repeated at least once in 19 patients. UBOs were found in 11 subjects (35.5%). Of the 3 patients with intellectual disability, 2 displayed UBOs. UBOs decreased in 4 patients who received repetitive brain MRI monitoring for more than 5 years.
Lastly, 7 patients presented tumours (22.6%), including 3 optic pathway gliomas (Fig. 1) (1 was bilateral), 3 plexiform neurofibromas, and 1 pilocytic astrocytoma of the brainstem (Fig. 2). Tumours were detected as a result of the first MRI scan taken during childhood, except for 1 plexiform neurofibroma and the pilocytic astrocytoma, which were revealed by imaging tests 7 and 10 years later. In both cases, tumours were searched for intentionally because of the presence of neurological symptoms.


No age differences were observed concerning the presence or absence of café-au-lait spots, ephelides, Lisch nodules, skeletal changes, or tumours. Analysis of the variable ‘presence of UBOs’ revealed an age-related difference; the mean age was significantly lower in subjects presenting these lesions (22.45±8.22 years vs. 32.5±10.64; P=.011). On the other hand, the variable ‘neurofibromas’ also showed age differences; on average, subjects with neurofibromas were significantly older than subjects lacking them (30.56±10.68 years vs. 18.25±4.34; P=.032).
No sex differences were found for any of the study variables. Concerning patients’ referring department, 21 were diagnosed by paediatricians, 7 (with a documented family history of NF1) by a dermatologist, and 3 were diagnosed as a result of a neurological examination (2 because of migraines and 1 due to optic nerve glioma).
DiscussionThe most frequent clinical finding in our series was the presence of café-au-lait spots. Only 1 subject presented fewer than the 6 spots required for diagnosis. Some series have reported a decrease in the number of café-au-lait spots beginning at age 50. However, our study did not reach this conclusion, probably because it contained so few patients in that age group.
Cutaneous neurofibromas usually appear after preadolescence and affect most adults with NF1.7 Our series has corroborated that subjects without neurofibromas are younger, which indirectly implies that neurofibromas appear with age. This type of benign lesions does not usually undergo malignant transformation.8 The frequency we report of inguinal and axillary freckling, 58%, is similar to that in other series.9 Lisch nodules are bilateral benign hamartomas of the iris that are pathognomonic to NF1. Researchers do not know if their incidence increases with age.9 Diagnosis of Lisch nodules requires a slit-lamp examination carried out by an ophthalmologist familiar with NF1.
As in other series, scoliosis, which affects one-sixth of these patients,10 appears during childhood, is idiopathic, and does not usually require surgery. Other skeletal changes typical of NF1 appeared only sporadically in our series, such as the single cases of sphenoid dysplasia and macrocrania.
We would like to highlight that mild intellectual disability was present in 3 of our patients (10%), and that UBOs were found in most of this subgroup. Although the small sample size does not allow us to draw conclusions, other authors have already made a connection between presence of UBOs and cognitive performance.11 The presence of neurological symptoms such as migraine and epilepsy seems to reflect isolated associations,7 considering that their prevalence in our group is similar to that in the general population (16% and 3% respectively). Other studies have related the presence of epilepsy to intracranial lesions, but not to UBOs.12 The only patient with seizures in our study presented an ischaemic lesion caused by Moyamoya syndrome.
The presence of UBOs in MRI scans was 35.5% lower than that in other (paediatric) series.5,12,13 Cross-sectional studies have associated the presence of UBOs with different forms of NF1, such as optic gliomas, Lisch nodules, and neurofibromas.14 Unlike neurofibromas, which seem to increase with age, UBOs gradually disappear.5,15 Data from our analysis support these findings, given that UBO prevalence was lower in our adult series and we also identified an age effect; subjects with UBOs were younger than those without these lesions.
The frequency of brain tumours in our series resembles that in earlier series; optic nerve gliomas were found in approximately 10% and pilocytic astrocytomas of the brainstem were found in 3%.16,17 Although the MRI scans showed a decrease in UBOs in several patients, we cannot draw conclusions since follow-up was irregular and numerous older neuroimaging tests are lacking. However, optic gliomas and the plexiform neurofibroma in our series have remained stable over time. The patient with stenosis of the aqueduct of Sylvius presented an optic nerve glioma which required a ventriculoperitoneal shunt 5 years later. This association has frequently been described in children with NF1.18 Although brain tumours in NF1 are usually benign, doctors should suspect malignancy in patients whose tumours are in an unusual location or behave in an uncharacteristically aggressive manner.19
Current consensus holds that NF1 leads to tumours not only in the peripheral nervous system (neurofibromas, etc.) but also in the central nervous system. The higher incidence of tumours in NF1 compared to the general population suggests that a brain MRI should be performed in NF1 patients with neurological symptoms, even if the age limit for serial studies remains unclear. We suggest that patients with UBOs undergo imaging studies in order to track any decreases in lesion size over time. MRI studies should also be performed in cases of focal neurological or ophthalmological signs.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Jiménez Caballero PE, et al. Manifestaciones clínicas y neurorradiológicas en los adultos con neurofibromatosis tipo 1. Neurología. 2013;28:361–5
