



Andersen-Tawil syndrome is a clinical entity characterised by periodic paralysis, distinctive physical features, and electrocardiographic alterations. This rare disease is frequently difficult to diagnose; the long exercise test may be useful in differential diagnosis including other muscle channelopathies.1–7 Several alterations have been described in the KCNJ2 gene, which encodes an inwardly rectifying potassium protein of the muscle membrane. We present the case of a family with the R67W mutation of KCNJ2.
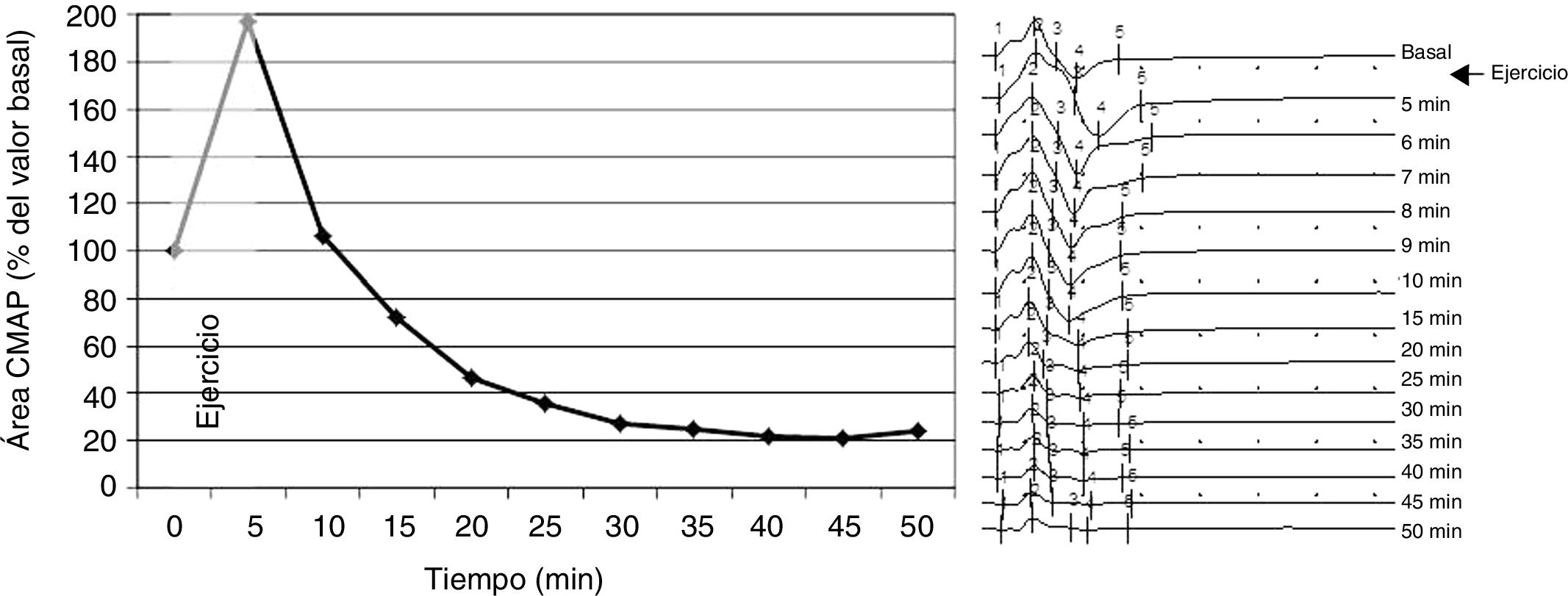
Our patient was a 39-year-old man who began to present episodes of generalised weakness at the age of 7-8 years; episodes lasted approximately 2 weeks, during which the patient was unable to leave his bed. Until the age of 25, episodes were frequent (presenting every month) and were exacerbated by carbohydrate and alcohol consumption, intense exercise, and rest after exercise. Such foods as bananas and potatoes did not trigger the episodes. Elevated serum potassium levels were detected at onset of the disease, leading to a diagnosis of hyperkalaemic periodic paralysis. As the disease progressed, episodes became less frequent, and some were associated with normal or even low potassium levels. The patient was referred to the neurophysiology department for evaluation due to mild generalised weakness of several months’ progression. He presented elevated CPK levels at admission (1325 U/mL). We performed a neurophysiological study including the short and long exercise tests, according to the protocol described by Fournier et al.1 The short exercise test was performed 3 times, with a 1-minute rest period between trials; results were normal. The long exercise test revealed a baseline amplitude of 10.7 mV, with a marked increase (197%) in the area of the compound muscle action potential (CMAP) after effort, followed by a progressive decrease until a minimum value of 21% was reached at 45 minutes (Fig. 1). Repetitive stimulation yielded normal results. The clinical progression, neurophysiological findings, and mild but characteristic phenotype (short stature, hypertelorism, micrognathia, and clinodactyly) observed in our patient led us to suspect Andersen-Tawil syndrome. The ECG revealed no ventricular arrhythmia; QT interval was within normal values. A genetic study detected a heterozygous mutation in the KCNJ2 gene (nucleotide change 199C > T, resulting in the substitution of arginine for tryptophan [R67W]). A genetic study of the patient’s relatives detected the mutation in his mother, but not in his sister (the only sibling). Our patient’s mother underwent the long exercise test, with normal results. She presented the same dysmorphic features as her son and ventricular extrasystoles, but had no muscle symptoms.
 Graphical representation of the progression of the CMAP area. B) Different CMAPs obtained during the test. Sensitivity 10 mV, sweep 50 ms, stimulus intensity 27 mA.")
Muscle channelopathies are rare diseases whose diagnosis is often challenging. In many cases, clinical symptoms are not sufficient to determine the type of muscle channelopathy, particularly when physicians are not familiar with this group of disorders; Andersen-Tawil syndrome is associated with mild phenotypes (some patients may even display no dysmorphic features). Potassium levels may be misleading, as in the case presented here, and genetic studies are costly. The long exercise test is easy to perform and may contribute valuable diagnostic data. Several patterns have been described,1–7 with the most frequent finding being a decrease of over 40% in CMAP; this finding was also observed in our patient and was key to diagnosis. Our patient displayed a marked increase in CMAP after prolonged effort, followed by a progressive decrease. In 2016, Song et al.3 described 12 patients with Andersen-Tawil syndrome who displayed 4 different patterns in the long exercise test; one pattern was characterised by a marked increase in CMAP amplitude during exercise, followed by a rapid decrease after exercise. Tan et al.4 and Díaz-Manera et al.5 described a slow decrease in CMAP after prolonged muscle contraction, without a significant increase in CMAP during the test. The pattern observed in our patient has not previously been described in the literature. Although our patient’s results in the short exercise test were normal, some patients with Andersen-Tawil syndrome have shown alterations in both the short and the long exercise test.5
Several mutations in the KCNJ2 gene have been described to date. Mutation R67W had previously been detected in a large cohort8 and was associated with a sex-specific phenotype, with muscle weakness and no cardiac alterations in men, and cardiac alterations but no muscle weakness in women; however, these patients did not perform the long exercise test. We hypothesised that our patient’s mother may have had subclinical muscle alterations, but the long exercise test yielded normal results.
In the case presented here, the test was extremely useful for 2 reasons: it guided diagnosis before the genetic study was conducted, and it provided objective data to further support the association between sex and phenotype in patients with Andersen-Tawil syndrome and the R67W mutation.
FundingThis study has received no public or private funding.
Please cite this article as: Parra S, Leal D, Vilar R, Mateo M, Melero J. Síndrome de Andersen-Tawil con fenotipo sexo-específico: utilidad del test de ejercicio largo. Nefrologia. 2020;35:675–676.
