



Ethylmalonic encephalopathy (EE) is an extremely rare disease caused by a recessive defect of the ETHE1 gene; it presents in infancy and follows a progressive course characterised by psychomotor retardation, hypotonia, and generalised microvascular damage. Delayed growth with diarrhoea and dysphagia is a common symptom. Neurological impairment is accelerated in the context of intercurrent infectious diseases, and patients typically do not live beyond the first decade of life.1,2
Diagnosis is based on clinical and laboratory findings. Patients with EE frequently present elevated levels of lactic acid, C4- and C5-acylcarnitine, and thiosulphate in the blood, and ethylmalonic acid in the urine. Diagnosis is usually confirmed in genetic studies.3,4 Treatment is currently symptomatic.5
We present the clinical, biochemical, radiological, and genetic data of a new patient with EE who was diagnosed during an infectious decompensation, and review the available evidence on this rare entity and its management.
The patient was a boy of 1.3 years of age with no relevant family or perinatal history who presented developmental delay, mild language regression with the loss of the 3 words he had learnt, hypotonia, and failure to thrive since the age of 10 months. He also presented capillary fragility, manifesting as petechiae on the limbs.
He was admitted to hospital due to acute decompensation in the context of fever and diarrhoea, with metabolic acidosis (pH, 7.21; pCO2, 14 mm Hg; HCO3, 8.8 mmol/L), hyperlactacidaemia (3.8 mmol/L), hyperglycaemia (glucose, 264 mg/dL), and ketonaemia (3.8 mmol/L); the patient was transferred to the intensive care unit for stabilisation. Examination at the paediatric ward revealed body weight of 7.4 kg (< p1; –3.2 SD), length of 72 cm (< p1; –3.24 SD), head circumference of 45.5 cm (p12; –1.18 SD), and a phenotype with dolichocephaly, prominent forehead, retrognathia, low-set ears, and a small mouth with thin lips. He presented petechiae, mainly on the forearms and popliteal and antecubital fossae. Interaction was good, with scarce language (2 or 3 doubtfully referential disyllables), good visual tracking, bilateral grasping, and marked global hypotonia; the patient was able to sit with support but could not stand nor walk. He was unable to push up to hands and knees from a prone position, and presented generalised hyperreflexia, sustained clonus, and bilateral Babinski sign.
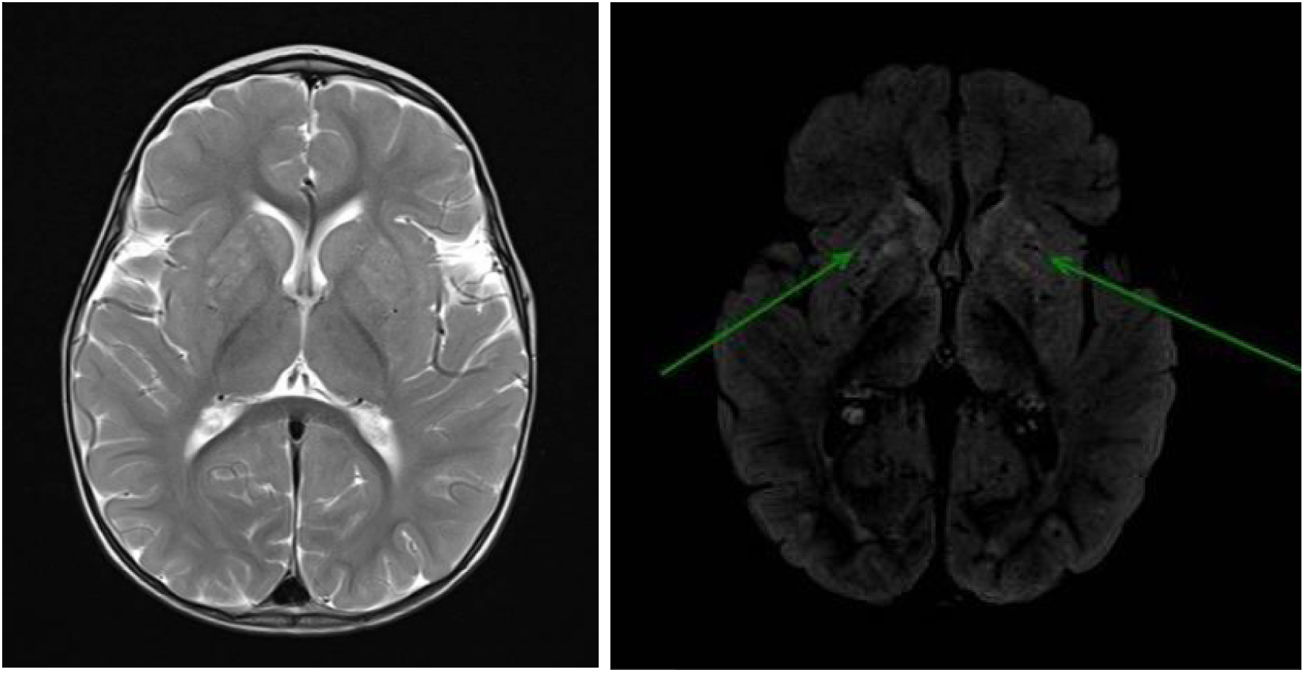
Brain MRI (Fig. 1) revealed signal alterations in the putamen and the head of the caudate nucleus bilaterally.
 and FLAIR sequences (right: arrows). No globus pallidus or internal capsule involvement were observed. No loss of parenchymal volume was observed at the supra- or infratentorial levels.")
Brain MRI scan: bilateral signal alterations in the putamen and head of the caudate nucleus; hyperintense lesions on T2-weighted (left) and FLAIR sequences (right: arrows). No globus pallidus or internal capsule involvement were observed. No loss of parenchymal volume was observed at the supra- or infratentorial levels.
The metabolic study of amino acids, blood acylcarnitines, and urine organic acids at 24 hours revealed elevated excretion of ethylmalonic acid. A targeted genetic panel revealed a homozygous pathogenic mutation of the ETHE1 gene (c.488 G > A [p.Arg163Gln]); both parents were asymptomatic carriers of the mutation.
Treatment was started with biotin, coenzyme Q10, vitamin E, riboflavin, thiamine, and L-carnitine due to suspicion of mitochondrial disease; metronidazole and N-acetylcysteine were subsequently added. Two months after admission, we observed considerable improvements in nutrition and in psychomotor development. We continued treatment with a low-protein diet, coenzyme Q10, riboflavin, L-carnitine, metronidazole, and N-acetylcysteine, which stabilised the patient, although failure to thrive and developmental delay (predominantly in motor areas) persisted. At the age of 2.5 years, he was able to walk with the assistance of one hand, uttered words but not sentences, and showed good interaction with his surroundings.
The genetic basis of EE was first described in 20041. The ETHE1 gene, located on chromosome 19, encodes a metallo-β-lactamase involved in the mitochondrial pathway, which is needed for the catabolism of hydrogen sulphide (H2S). This defect causes accumulation of H2S and its derivatives (thiosulphate) in various fluids and tissues, and induces direct damage to cell membranes and inhibits cytochrome c oxidase and short-chain acyl-CoA dehydrogenase. It increases levels of lactic acid, ethylmalonic acid, and C4- and C5-acylcarnitine.1,5,6 Various mutations have been identified, the majority originating in the Mediterranean and the Middle East,2,6 with variability between families.4,7,8 The characteristic mucocutaneous manifestations are caused by microvascular toxicity and include recurring petechiae, cutis marmorata, haemorrhagic mucosal suffusion, and/or distal orthostatic acrocyanosis.1,8 Hydronephrosis, cryptorchidism, and cardiac anomalies have also been described.7
The typical brain MRI alterations are irregular contrast-enhancing hyperintense lesions in the basal ganglia,7,9 as in our patient, resembling those involved in Leigh syndrome. Other abnormalities described in the literature include hyperintensities in the brainstem, cortical atrophy, diffuse leukoencephalopathy, and congenital malformations such as tethered spinal cord and Chiari malformation.2
Gene sequencing is recommended for diagnosis. If pathogenic variants are not identified, or are found in a single allele, then targeted deletion and duplication analysis of the gene should be performed.3,10
Treatment should aim to guarantee adequate nutritional support.2 Treatment with riboflavin, L-carnitine, and coenzyme Q10, as administered in our patient, seems beneficial.11 Diets with restricted intake of sulphurated amino acids also improve symptoms and biochemical markers.5 Combined use of metronidazole and N-acetylcysteine has been shown to control accumulation of H2S by reducing the abundance of the bacteria producing sulphide and neutralising H2S, respectively.12,13 These patients occasionally require renal replacement therapy.14
Liver transplantation seems to be a therapeutic option,9 and gene therapy with adeno-associated viral vectors is currently under study.15
Finally, it should be noted that the phenotype associated with the c.488 G > A (p.Arg163Gln) mutation of ETHE1 behaves as a mitochondrial disease, associated with capillary fragility and failure to thrive, and responds well to mitochondrial stimulation, antioxidants, and gastrointestinal decontamination.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Cardelo Autero N, Cordón Martínez AM, Ramos-Fernández JM. Encefalopatía etilmalónica: descripción fenotipo-genotipo y revisión de su manejo. Neurología. 2021;36:729–731.
