



Mir-146a-5p has been widely recognized as a critical regulatory element in the immune response. However, recent studies have shown that miR-146a-5p may also be involved in the development of Alzheimer disease (AD). Regrettably, the related mechanisms are poorly understood. Here, we investigated the effects of miR-146a in mice models and SH-SY5Y cells treated with amyloid β (Aβ)1–42.
MethodsTo create a model of AD, SH-SY5Y cells were treated with Aβ1–42 and mice received intracerebroventricular injections of Aβ1–42. Then, the transcriptional levels of miR-146a were estimated by real-time PCR. We transiently transfected the miR-146a-5p mimic/inhibitor into cells and mice to study the role of miR-146a. The role of signaling pathways including p38 and reactive oxygen species (ROS) was studied by using specific inhibitors. Aβ and amyloid-beta precursor protein (APP)levels were measured by immunoblotting. Furthermore, Aβ expression was analyzed by immunofluorescence and histochemical examinations.
ResultsAβ1–42-stimulated SH-SY5Y cells displayed increased transcriptional levels of miR-146a and APP. Moreover, the p38 MAPK signaling pathway and ROS production were activated upon stimulation with a miR-146a-5p mimic. However, treatment with a miR-146a-5p inhibitor decreased the levels of APP, ROS, and p-p38 MAPK. A similar phenomenon was also observed in the animals treated with Aβ1–42, in which miR-146a upregulation increased the expression of Aβ, p-p38, and ROS, while the inhibition of miR-146a had the opposite effect. This suggests that miR-146a increases Aβ deposition and ROS accumulation via the p-p38 signaling pathway.
ConclusionsOur research demonstrates that miR-146a-5pa increases Aβ deposition by triggering oxidative stress through activation of MAPK signaling.
miR-146a-5p es un elemento regulador clave en la respuesta inmune. Sin embargo, estudios recientes sugieren que miR-146a-5p también está involucrado en el desarrollo de la enfermedad de Alzheimer (EA), aunque aún no se conoce con exactitud el mecanismo por el que esto sucede. Analizamos los efectos de miR-146a en un modelo animal y en células SH-SY5Y expuestas a β-amiloide (Aβ)1-42.
MétodosTratamos células SH-SY5Y con Aβ1-42 e inyectamos Aβ1-42 en los ventrículos cerebrales de ratones para generar un modelo celular y otro animal de EA. Estimamos los niveles transcripcionales de miR-146a mediante PCR en tiempo real. Al mismo tiempo, transfectamos temporalmente las células y los ratones con imitador/inhibidor de miR-146a-5p para evaluar el papel de miR-146a. Estudiamos el papel de algunas vías de señalización, como la de p38, y los niveles de especies reactivas de oxígeno (ERO) con inhibidores específicos. Los niveles de Aβ y de proteína precursora amiloidea (APP) se determinaron con inmunoblot. También se analizó la expresión de Aβ mediante inmunofluorescencia y análisis histoquímico.
ResultadosLas células SH-SY5Y expuestas a Aβ1-42 mostraron altos niveles transcripcionales de miR-146a y APP. La vía de señalización p-38 MAPK y la producción de EROs se activaron al utilizar un imitador de miR-146a-5p. Sin embargo, el bloqueo de miR-146a-5p con un inhibidor redujo los niveles de APP, EROs y p-p38 MAPK. Se observó un fenómeno similar en los ratones tratados con Aβ1-42: la sobrerregulación de miR-146a aumentó la expresión de Aβ, p-p38 y EROs, mientras que la inhibición de miR-146a tuvo el efecto contrario. Esto sugiere que miR-146a está involucrado en el aumento de acumulación de Aβ y de producción de EROs por medio de la vía de señalización p-p38.
ConclusionesNuestro estudio muestra que miR146a-5p aumenta la acumulación de Aβ al promover el estrés oxidativo a través de la activación de la vía de señalización MAPK.
Alzheimer's disease (AD), the most common degenerative disorder of the nervous system, exhibits typical clinical pathological changes linked to the dysfunction of cognitive ability.1 The pathogenesis of AD is an extremely complicated process, of which multiple hypotheses have been proposed. The most usual is the amyloid deposition hypothesis which suggests the accumulation of the neurotoxic Aβ fragments derived from amyloid precursor protein (APP) by the action of β-secretase and γ-secretase proteases.2,3 Recently, a study revealed that Aβ deposition in the brain triggers oxidative stress, affecting the reduction ability of mitochondria, causing neuronal death.4 Besides, antioxidants are known to reverse neuronal cell apoptosis induced by Aβ.5 Therefore, these studies emphasize that oxidative stress is a significant factor in Aβ-triggered neuronal death, which gradually may turn to AD.
MicroRNAs (miRNAs), a kind of small non-coding RNAs, are known to play a major post-transcriptional regulatory role in gene expression. Recently, the involvement of many miRNAs in modulating key disease genes, such as APP and BACE1, suggested that their dysfunction may also contribute to the pathology of AD.6 miR-146a-5p, the most widely studied miRNAs, is the key modulator of immune response and has been linked to a variety of neuroinflammation processes.7 The up-regulation of miR-146a-5p activates mitogen protein kinase to exacerbate neuroinflammation and oxidative stress.8 Interestingly, studies based on in vitro and in vivo models of AD found that gradual up-regulation of miR-146a-5p was linked to the progression of AD, while the other studies suggested that miR-146q-5p was closely related to Aβ deposition and synaptic pathological changes.9 Meanwhile, clinical studies also revealed that compared to the healthy elderly group, miR-146a-5p was significantly up-regulated in the brain tissue of AD patients.10 In general, both preclinical and clinical trials strongly put forward the role of miR-146a-5p in the pathogenesis of AD. Therefore, we conjectured that regulating miR-146a-5p could be a novel therapeutic strategy in AD.
However, for that, the mechanistic details of miR-146a-5p role in oxidative stress-induced neuronal degeneration in AD need to be examined. Therefore, in this study, we evaluated whether the down-regulation of miR-146a-5p would cease the progression of AD. Also, we discussed the relevant pathological changes in AD resulting from the down-regulation of miR-146a-5p.
Material and methodsCell culture, transfection, and Aβ1-42 treatmentSH-SY5Y cells were obtained from Procell (No.CM-0208, China) and cultured in MEM/F12 medium (Procell, No.PM151220, China) (Israel Biological Industries) with10% fetal bovine serum (FBS) at 37°C. To induce cell differentiation, MEM/F12 medium with 1% FBS and 10μM all-trans retinoic acid (RA) (Aladdin, No.CFLD-R106320, China) was used for 7 days, and the culture medium was changed every 3 days. After the RA treatment, the resultant morphological changes in SH-SY5Y cells were verified microscopically (200x magnification), and the differentiated cells were used for all subsequent studies. For transfection studies, SH-SY5Y cells were transfected with miR-146a-5p mimic (50nM; RiboBio, No.miR10000449-1-5, China) or miR-146a-5p inhibitor (100nM; RiboBio, No.miR20000449-1-5, China) using Lipofectamine3000 (Invitrogen, No.L3000-015, USA) according to the manufacturer's instructions. After incubation for 24h, these cells were harvested and undergoing further tests, meanwhile, the transfection efficiency also was monitored. To establish the in vitro AD cell model, Aβ1–42 (GenScript, No.RP10017, USA) was dissolved in hexafluoroisopropanol (HFIP) for 10min. The HFIP was pre-cooled in advance and after volatilization, the formed Aβ1–42 protein film was dissolved in DMSO, and the SH-SY5Y cells were treated with 1μM Aβ1–42 for 24h. To evaluate the effect of MAPK signaling, the SH-SY5Y cells were divided into the following groups with three replicates in each: (1) the control group without treatment, (2) the model group with Aβ1–42 treatmen, (3) the Model+miR-146a-5p mimic group with miR-146a-5p mimic transfection and Aβ1–42 treatmen, (4) the Model+miR-146a-5p mimic+ FGA-19 group was treated with Aβ1–42 to establish the in vitro AD cell model, and transfected with miR-146a-5p mimic, then FGA-19 (Aladdin, No.5.30486.0001, China) as the p38 MAPKinase inhibitor was added in the cells for 24h with concentration of 50μM, (5) the Model+miR-146a-5p mimic+ FGA-19+NAC group was treated similarly to the Model+miR-146a-5p mimic+ FGA-19 group, in addition, the cells were treated with N-Acetyl-cysteine (NAC, Abcam, No.AB60264, USA) of 1mM for 2h, as antioxidant to scavenge ROS.
Animals and treatmentThe animal experiment was approved by the Animal Ethics Committee of the Affiliated Hospital of Chengde Medical College (No.20200330-06). All animal studies strictly complied with the relevant regulations of the Animal Ethics Committee and abided by the 3R principle in the design and implementation of experiments. Six-month-old male C57bl/6J mice were randomly assigned into four groups (n=5 per group), namely the control group without treatment, the AD group with Aβ1–42 treatment, the AD+miR-146 mimic group with miR-146a-5p mimic transfection and Aβ1–42 treatment, and the AD+miR-146 inhibitor group with miR-146a-5p inhibitor transfection and Aβ1–42 treatment. The lentiviral expression vectors of miR-146a-5p mimic, miR-146a-5p inhibitor and negative control were synthesized by Thermo Scientific company with their titer of 1×109PFU/mL. Except for the control group, the other three groups of animals were intracerebroventricular injected with Aβ1–42 which was prepared by dissolving in distilled water at 0.2mg per mL. On contrary, the animals in the control group received the same volume of distilled water (vehicle control). Moreover, the mice in the AD+miR-146 mimic group were also intracerebroventricular injected with 3μL lentiviral expression vector of miR-146a-5p mimic, the treatment in the AD+miR-146 inhibitor group was similar. After two weeks of the treatment period, the animals were examined for cognitive behavior using the Morris water maze (MWM) test. After that, all the mice with five in each group were anesthetized and sacrificed to obtain the hippocampal tissues for further study. Besides, three sections of hippocampal tissues per mouse were analyzed.
Surgical procedureThe mice were placed in the isoflurane anesthesia device, adjusted to scale 2, with a 400cc/min air flow rate. Post-anesthesia, the mice were quickly fixed to the brain stereotaxic device and a wound was cut with a sterile scalpel blade. The brain surface was disinfected using an cotton swab dipped with alcohol. Then, an insertion point (coordinate: ML±1.0; AP 0.3) with 0.5mm diameter aperture was drilled with a hand drill using a stereotactic instrument, and Aβ1–42 was injected to establish the animal model of AD.
Morris water maze (MWM)MWM behavioral experiment was performed on the 14th day after the operation. In the experiment, a circular pool (50cm high and 20cm in diameter) was filled with opaque water (22±3°C), and a circular platform (10cm in diameter, 28cm high) was placed 1cm below the surface of the water. The MWM was theatrically divided into four quadrants, and the hidden platform was placed in one of the quadrants. The experiment was divided into two stages. Stage one, the first 5 days was used for the positioning and navigation, in which each mouse was tested 4 times a day. In each test, the mice were placed in the water from different quadrants and allowed to swim for up to 90s to find the platform and rest on it for 10s. If the mouse failed to find the platform within the specified time, it was guided to the platform and stand for 10s, meanwhile, the escape latency was recorded as 90s. In the second stage of free exploration, the platform was hidden on the 6th day. Then, the swimming time, distance of the mouse in the quadrant before the platform within 90s, and the number of times it crosses the platform was recorded. The acquired data was analyzed.11
Estimation of cellular reactive oxygen species (ROS) generationAccording to commercial regulations, the DCFH-DA method was used to estimate the production of cellular ROS. DCFH-DA is a fluorescent dye detecting ROS level, which can be converted to oxidized fluorescence dye 2’,7’-dichlorofluorescein (DCF) at the presence of ROS. First, SH-SY5Y (4×104) cells were seeded into a 96-well plate and exposed to Aβ1–42 for 1day (25μM). After this, the cells were incubated for 30min in the DMEM medium containing 5μ DCFH-DA (MedChemExpress, No.HY-D0940, USA) under dark conditions at room temperature (RT), and the cells were washed with PBS. Moreover, the nucleus with DAPI (Bio-Rad, No.1351303, USA) to distinguish apoptotic cells. Fluorescence images were captured immediately using a fluorescence microscope (Zeiss Axio Imager Z2, Germany) at 10× magnification.
Aβ1–42 ELISAAβ1–42 analysis was performed according to the Aβ1–42 ELISA kit (Invitrogen, No.KHB3441, USA) operating instructions. Cell homogenates of the hippocampal tissue were prepared. Meanwhile, Aβ standard solution was prepared and the test samples were diluted with the standard dilution buffer provided in the kit. Then, 100μL of standards was added to the appropriate microtiter wells in triplicates and incubated overnight at 4°C. The next day, the liquid in the 96-well plate was completely removed and washed 3 times with washing buffer. Then, the Aβ1–42 antibody was added to the sample and incubated for 60min at 37°C. Again, 3 times washing was performed with the cleaning solution and incubation with the secondary antibody was carried out for 30min at RT. Once again, after washing each well at least 3 times, 100μL of stable color developing solution was added to each well. Lastly, the absorbance was recorded at 450nm to calculate the concentration of Aβ1–42 in the corresponding samples using the standard curve.12 The antibodies and solutions were provided in the kit.
ImmunohistochemistryUsing the cryostat, coronal sections (thickness: 40μm) of brain tissue were obtained. These were washed 3 times with 1% PBS for 5min and then incubated with 5% bovine serum albumin in an incubator at RT for 0.5h. Next, these were incubated with primary antibodies (in PBS, Anti-Aβ1–42; 1:1000; Invitrogen, No.MA5-36246, USA) at RT for 60min, and then overnight at 4°C. After overnight incubation, tissue sections were bought to RT and washed 3 times with 1% PBS for 5min. Followed by incubation with the secondary antibodies (goat anti-rabbit; 1:1000; Invitrogen, No.A32731, USA) at 37°C for 1h, the brain slices were washed 3 times with 1% PBS for 5min. Lastly, the brain tissue sections were dipped in the DAB (Sigma, No.11718096001, USA) chromogenic solution with the deposition of Aβ1–42 stained dark brown and mounted on glass slides. After dried in a 37°C incubator, tissue sections were dehydrated using ethanol gradient and turned transparent with xylene. Finally, the images were acquired using an optical microscope.13
RNA extractionThe total RNA of each sample was extracted by the RNAiso Plus Reagent (Takara, No.M9108, Japan). The frozen cells were lysed using TRIZOL and placed at RT for 5min for completed isolution. Then, 0.2ml of chloroform was added to every 1ml of the lysed sample and mixed vigorously for 15s. The mixture was incubated at 15–30°C for 2–3min and then centrifuged at 12,000rpm for 15min at 4°C. After centrifugation, the RNA, distributed in the water phase, was precipitated using an equal volume of isopropanol. After the precipitation, the RNA pellet was rinsed with at least 1ml of 75% ethanol (75% ethanol prepared with DEPCH2O) and re-centrifuged at 7000rpm at 4°C for 5min. Next, most of the ethanol solution was carefully removed and the RNA pellet was air-dried at RT for 5–10min. Lastly, The RNA pellet was dissolved in 40μl of RNase-free water and stored at −80°C for later use.
qRT-PCR AssayFor qRT-PCR, cDNA synthesis was performed according to the manufactures’ instruction for the PrimeScriptTM RT Master Mix Kit (Takara, No.RR036B, Japan). The reverse transcriptase MMLV along with the reaction mixture was incubated at 70°C for 3min, and then immediately transferred to ice water. Then, 0.5μl of reverse transcriptase was added and incubated at 37°C for 60min. Next, final incubation was performed at 95°C for 3min to obtain the cDNA which was stored at −80°C. The housekeeping gene, β-actin was used as an internal standard. The specific primers used were: for miR-146a-5p, 5’-3’ (forward) GGG GTG AGA ACT GAA TTC CAT and 5’-3’ (reverse) CAG TGC GTG TCG TGG AGT; for β-actin, 5’-3’ (forward) TGG CAC CCA GCA CAA TGA A and 5’-3’ (reverse) CTA AGT CAT AGT CCG CCT AGA AGC A. The target gene and housekeeping gene of each sample were designed and synthesized by Shanghai GenePharma Company (China), and subjected to real-time PCR by means of the SYBR@Premix Ex TaqTM (Tli RNaseH Plus) Kit (Takara, No.RR820A, Japan). Real-time PCR was performed with the following cycling conditions: 95°C for 30s, 40 cycles of 95°C for 5s, 60°C for 30s and, after that, 95°C for 15s, 60°C for 1min, 95°C for 15s. PCR products were electrophoresed on a 2% agarose gel, and stained with GoldView™ (Shanghai yuanye Bio-Technology, No.R20977, China) to detect the amplified product. The relative expression level was calculated using the 2-ΔΔCT method.
Western blottingSH-SY5Y cells and hippocampal tissue were lysed with RIPA buffer (Abcam, No.AB156034, USA) under specific conditions and separated on SDS polyacrylamide gel (Abcam, No.AB139597, USA). The protein bands were then transferred onto a PVDF membrane (Sigma, No.3010040001, USA) which was blocked with 5% skimmed milk. After blocking, three times washing was performed with PBS. Then, the membrane was incubated overnight with the primary antibodies, APP antibody (1:500, Abclonal, No.A11912, China), Aβ1–42 antibody (1:200, Abcam, No.P05067, USA), p38 MAPK antibody (1:500, Abcam, No.AB170099, USA), p-p38 MAPK antibody (1:1000, Beijing Biolab, No.K22589-TZH, China) or GADPH antibody (1:5000, Cell Signaling Technology, No. 4967, USA) at 4°C. On the second day, after rewarming for 30min and 3 times washing with PBS, membranes were incubated with the corresponding HRP-conjugated goat anti-rabbit IgG or HRP-conjugated goat anti-mouse IgG (1:10,000, Sigma, No.A0545 and SAB3700986, USA) secondary antibodies for 30min at RT. The membranes were again washed 3 times with PBS. Lastly, the ECL reagent (Sigma, No.WBULS0500, USA) was used to illuminate the target protein bands, and the quantitative analysis of protein was performed by a Gel-Pro-Analyzer imaging system.14
Statistical analysisAll experimental results are presented as mean±standard error, and P<0.05 denotes the significant statistical difference. For statistical analysis, Student's t-test (comparison between two groups) and ANOVA test (comparison between multiple groups) were used. Also, Bonferroni correction was used for the post hoc test. Two-way ANOVA and repeated measures were used to determine the time differences between the two groups in the MWM and the group factors (based on the escape latency). The software SPSS version 21.0 was used for the statistical analysis.
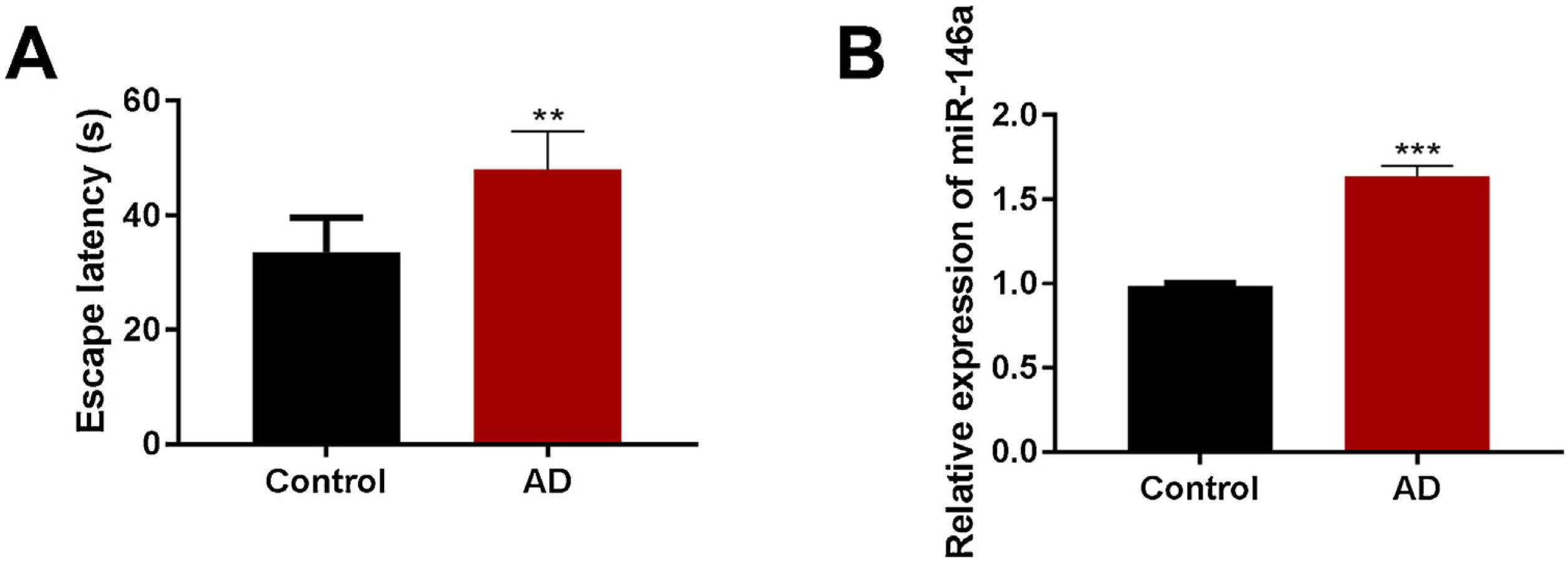
ResultsUpregulated miR-146a-5p elevated cognitive impairment in Aβ1–42-treated micemiR-146a-5p is known to be involved in the pathogenesis of AD. In our study, we observed that the transcription of miR-146a-5p was significantly up-regulated in the Aβ1–42-treated group compared to the control group (Fig. 1B). Also, Aβ1–42-treated mice exhibited higher escapes latency than the mice of the control group (Fig. 1A). Overall, these results indicated a strong positive correlation between cognitive impairment and increased levels of miR-146-5p in mice model of AD.
 Estimation of escape latency in Morris water maze in the control and Aβ1–42-treated groups (n=5); (B) Relative expression of miR-146a-5p in the control and Aβ1–42-treated groups (n=5). Each bar represents mean±S.E.M. * P<0.01 and ***P<0.001 vs. the control group.")
The expression of miR-146a-5p and cognitive impairment were elevated in Aβ1–42-treated mice. (A) Estimation of escape latency in Morris water maze in the control and Aβ1–42-treated groups (n=5); (B) Relative expression of miR-146a-5p in the control and Aβ1–42-treated groups (n=5). Each bar represents mean±S.E.M. * P<0.01 and ***P<0.001 vs. the control group.
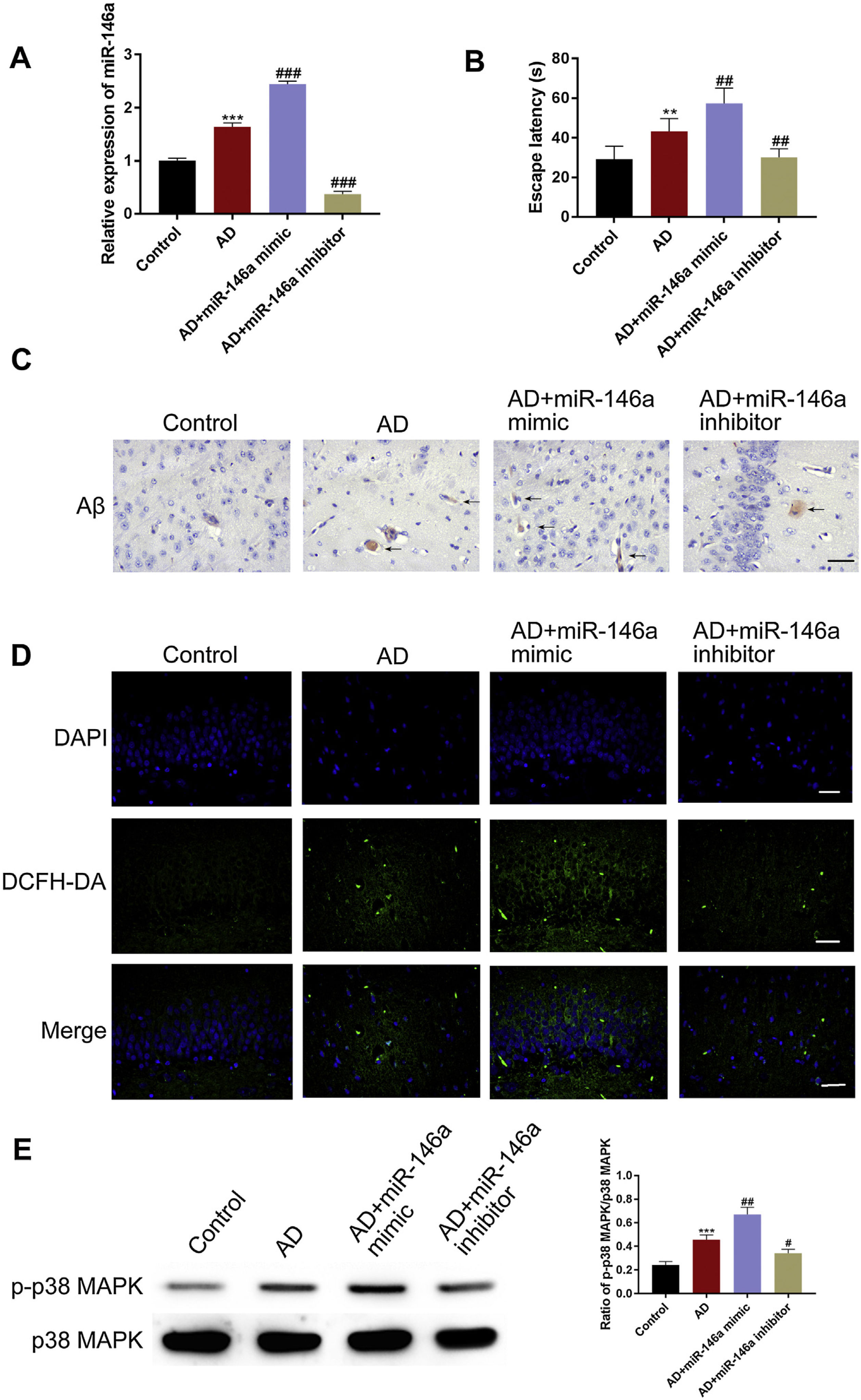
Next, we investigated the correlations between cognitive impairment and miR-146a-5p by using miR-146a-5p mimic and miR-146a-5p inhibitor. We first evaluated the cognitive ability among the groups. The MWM results showed that compared to the control group, the escape latency increased in the AD group, and was further aggravated in the miR-146a-5p-mimic group. However, the treatment with miR-146a-5p inhibitor markedly reversed this change (Fig. 2B). Also, when we examined the levels of miR-146a-5p and Aβ deposition, we observed that compared to the control group, the levels were significantly higher in the model group. Notably, the miR-146a-5p expression and Aβ deposition were further increased in the miR-146a-5p-mimic group. However, these levels were lowered upon treatment with miR-146a-5p-inhibitor (Fig. 2A and C). Next, we tested the role of MAPK signaling and ROS level. We observed that compared to the control group, a greater level of p-P38 was observed in the miR-146a-5p mimic group, and ROS were accumulated. However, a reverse was observed upon treatment with miR-146a-5p-inhibitor (Fig. 2D–E). These experiments suggested that miR-146a-5p promoted the deposition of Aβ by triggering oxidative stress via activation of the MAPK signaling pathway.
 Relative transcription levels of miR-146a-5p among groups (n=5); (B) Relative escape latency in MWM are shown (n=5); (C) Immunohistochemistry analysis of Aβ in the hippocampus upon treatment as indicated, Scale bar: 200μm; (D) Relative expression of DCHF-DA among groups, Scale bar: 50μm; (E) Bar graphs show densitometric analysis of p-p38as indicated in western blots (n=5). Each bar represents mean±S.E.M. * P<0.01 and *** P<0.001 vs. the control group. ##P<0.01 and ###P<0.001 vs. the AD group.")
miR-146a-5p promoted Aβ deposition by triggering oxidative stress via activation of MAPK signaling in Aβ1–42-treated mice. (A) Relative transcription levels of miR-146a-5p among groups (n=5); (B) Relative escape latency in MWM are shown (n=5); (C) Immunohistochemistry analysis of Aβ in the hippocampus upon treatment as indicated, Scale bar: 200μm; (D) Relative expression of DCHF-DA among groups, Scale bar: 50μm; (E) Bar graphs show densitometric analysis of p-p38as indicated in western blots (n=5). Each bar represents mean±S.E.M. * P<0.01 and *** P<0.001 vs. the control group. ##P<0.01 and ###P<0.001 vs. the AD group.
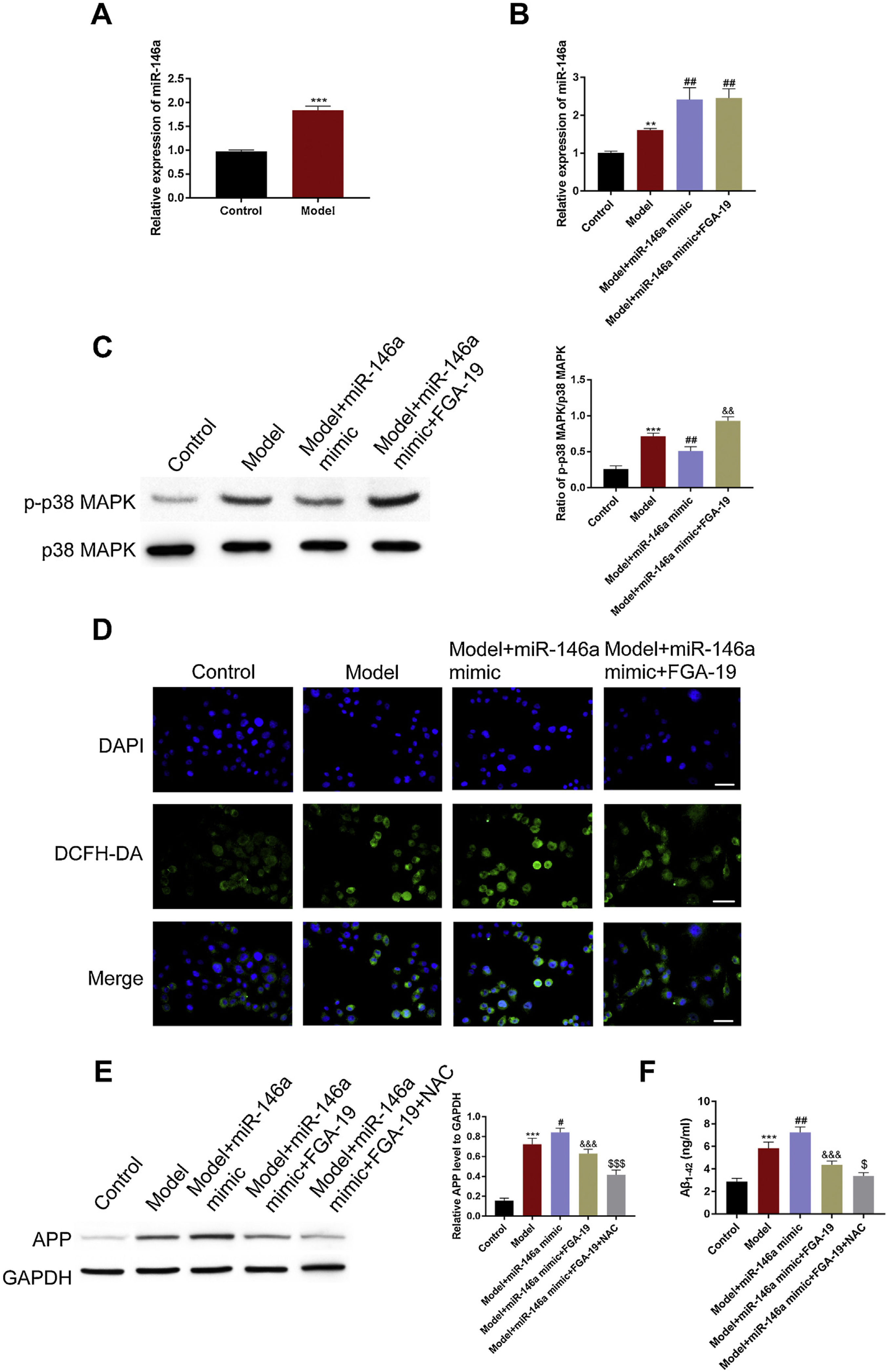
Next, we verified the aforesaid in vivo observations at the cellular level. For this, SH-SY5Y cells were used. Since miR-146a-5p is known to participate in the pathogenesis processes of AD, we began by evaluating the adverse effect of miR-146a-5p in the control and treated groups of cells. We observed that compared to the control group, Aβ1–42 treatment significantly up-regulated the level of miR-146a-5p (Fig. 3A) and APP (Fig. 3E). This suggested that the transcription levels of both miR-146a-5p and APP were influenced by Aβ1–42 treatment.
 Relative transcription levels of miR-146a-5p among groups (n=3); (C) Immunoblotting of p-p38 in the hippocampus following the treatment as indicated. (D) Relative expression of DCFH-DA among groups, Scale bar: 50μm; (E) Western blots showing the APP levels and the bar graphs show densitometric analysis of the same (n=3). (F) Bar graphs showing the densitometric analysis of Aβ1–42 by ELISA (n3). * P<0.01 and *** P<0.001 vs. the control group. ## P<0.01 and ### P<0.001 vs. the AD group.")
miR-146a-5p up-regulated APP by increasing ROS levels via activation of MAPK signaling in the Aβ1–42-treated SH-SY5Y cells. (A, B) Relative transcription levels of miR-146a-5p among groups (n=3); (C) Immunoblotting of p-p38 in the hippocampus following the treatment as indicated. (D) Relative expression of DCFH-DA among groups, Scale bar: 50μm; (E) Western blots showing the APP levels and the bar graphs show densitometric analysis of the same (n=3). (F) Bar graphs showing the densitometric analysis of Aβ1–42 by ELISA (n3). * P<0.01 and *** P<0.001 vs. the control group. ## P<0.01 and ### P<0.001 vs. the AD group.
ROS accumulation is an important pathological change in AD pathology and critical for the production of APP. To test whether the elevated level of APP was related to ROS, the DCFH-DA (a probe for indicating ROS) method was used. We found that ROS were immensely accumulated in the treated group rather than the control group (Fig. 3D). Next, we examined whether this was caused by miR-146a-5p. For this, we exploited miR-146a-5p mimic and miR-146a-5p inhibitor. We found that compared to the model group, in the miR-146a-5p mimic group, levels of miR-146a-5p, APP, Aβ1–42, and ROS were highly up-regulated (Fig. 3A, D, and F). Importantly, these effects were reversed in the miR-146a-5p inhibitor group. Meanwhile, we noticed that compared to the control group, the expression of p-P38 was elevated in the model group. Also, the miR-146-mimic group exhibited a higher level of p-p38 while the miR-146a-5p-inhibitor group displayed a lower level of p-p38 (Fig. 3C). Overall, these results indicated that the expression of APP and Aβ1–42 were influenced by miR-146 via activation of MAPK signaling and oxidative stress.
DiscussionMost degenerative diseases of the nervous system are closely related to age.15 AD is a disease of severe dementia with a growing incidence in humans. Understanding the pathogenesis at the cellular can reveal novel insights for the prevention and treatment of AD. In our study, we firstly verified that miR-146a-5p promoted the development of AD by depositing Aβ in the animal model. This was also true in cell assays. We think that the potential mechanism could be related to increased oxidative stress via activation of MAPK signaling.
miR-146a-5p is widely regarded as an inflammation-related microRNA with immunomodulatory effects.16 Multiple reports showed that miR-146a-5p could inhibit the interleukin-1 receptor-related kinase 1 (IRAK1) and down-regulate NF-κb activity in the cognitive and memory-related brain regions, such as the hippocampus and prefrontal cortex in AD model mice.17,18 Recently, other studies also implicated miR-146a-5p in aging processes using the experimental animal models of AD.19,20 From the postmortem brain autopsy of AD patients, studies found that miR-146a-5p was highly expressed in the CSF, serum, and plasma. Therefore, to verify the correlation between AD and miR-146a-5p, we first tested the level of miR-146a-5p in Aβ1–42 treated cells and animals. Notably, our findings are consistent with previous studies and found significantly higher levels of miR-146a-5p in the AD model group. Interestingly, we also found that reduced levels of miR-146 markedly decreased APP and deposition of Aβ in the hippocampus regions of the brain.
Several hypotheses put oxidative stress as the key factor in the pathophysiology of AD. With the accumulation of ROS, synaptic activity gradually decreases which leads to abnormal metabolism triggering the accumulation of Aβ and the hyper-phosphorylation of Tau protein. This ultimately causes mitochondrial dysfunction and neuronal cell death.5 The autopsy analysis of mouse models and AD patients revealed that an unbalanced redox state leads to increased ROS causing mitochondrial dysfunction and Aβ peptide aggregation.5,21 In our study, the ROS level were much higher in the model group than that of the control group, both in animal and cell studies, which also was consistent with the previous reports. Importantly, up-regulated miR-146a-5p further aggravated oxidative stress in the AD model group.
MAPKs, belonging to the class of protein Ser/Thr kinases, can transform extracellular stimuli into intracellular responses, and thereby regulate many physiological processes.22 ERK1/2, p38 MAPK, and c-Jun N-terminal kinases (JNKs) are the most widely studied MAPKs. Notably, MAPK signaling is also known to regulate the generation of ROS. In our study, the miR-146a-5p-mimic group exhibited abnormally increased levels of ROS, though this could be reversed by using FGA-19 (a MAPK inhibitor). The elevated Aβ levels were caused by oxidative stress and the unbalanced redox state was triggered by miR-146a-5p-induced MAPK signaling. These results strongly indicate that in the Aβ-treated cells and animal models, ROS production is dependent on the activation of the MAPK signaling pathway. Also, we think that miR-146a-5p inhibitors can be used to block MAPK signaling. In addition, there are some limitations should be noted in our study. For example, it indeed is difficult to differentiate the administer Aβ from the generated with our study design, which limits the research about relationship of miR-146a-5p and Aβ. In this study, the mice model of AD is induced by the intracerebroventricular injection of the Aβ1–42, however, there are relatively mature and widely used mice models of AD which can be purchased from company or research institution, and will be used directly in our future study.
ConclusionsIn conclusion, using the animal and cellular models, we reported that miR-146a-5p promotes the development of AD and aggravates Aβ deposition which is potentially caused by oxidative stress via activation of MAPK signaling.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestsThe authors declare that they have no conflict of interest.
