



The last fifteen years have seen the gradual appearance of a number of different drugs that have been shown to be effective as disease modifying therapies in multiple sclerosis. The opening and subsequent widening of the therapeutic armamentarium in multiple sclerosis will continue on a expanding course in the next few years due to the already known positive results of phase III clinical trials with orally administered molecules. Along with these, we have also seen the appearance of a group of drugs which, instead of being defined by their route of administration, are considered together as a consequence of their similar design: the monoclonal antibodies.
Contents and methodsThe principal safety and efficacy results of three of the monoclonal antibodies that have already obtained positive results in phase II studies will be reviewed in this paper: alemtuzumab, rituximab/ocrelizumab, and daclizumab. For the preparation of this paper, information was obtained from already published articles and from the following web pages: www.clinicaltrials.gov of the National Institute of Health of the USA, the EMA (European Medicines Agency) web page and the Spanish Medicines Agency (Agencia Española del Medicamento) web page.
ConclusionsFinal results from the phase III clinical trials in progress are required to produce definitive statements on the efficacy and safety of the reviewed drugs. However, and subject to confirmation of the presently available data from phase II trials, it is likely that this group of drugs is to be placed one step beyond the currently available disease-modifying therapies in terms of efficacy, but with a safety pattern which will make careful monitoring of treated patients a mandatory requirement so as to obtain adequate risk/benefit profiles.
Los últimos 15 años han visto aparecer de manera progresiva diferentes fármacos que se han mostrado eficaces para el tratamiento de fondo de la esclerosis múltiple. Esta inauguración y posterior ampliación del arsenal terapéutico en esclerosis múltiple seguirá un curso ascendente en los próximos años dados los resultados positivos ya conocidos de ensayos clínicos fase III con moléculas de administración oral. Junto a ellos también hemos observado la aparición de un grupo de fármacos que en lugar de definirse por su vía de administración lo hacen por su diseño: los anticuerpos monoclonales.
DesarrolloEn este artículo se revisan los principales resultados de seguridad y eficacia de tres de los anticuerpos monoclonales que ya han obtenido resultados positivos en estudios de fase II: alemtuzumab, rituximab-ocrelizumab y daclizumab. Para la elaboración de este trabajo se ha obtenido información de artículos ya publicados y de las siguientes páginas web: www.clinicaltrials.gov del National Institute of Health (NIH) de los EE. UU., de la EMA (Agencia Europea del Medicamento) y de la Agencia Española del Medicamento.
ConclusionesEs necesario disponer de los resultados de los ensayos fase III en marcha actualmente para emitir juicios adecuados sobre estos fármacos. Sin embargo, es de esperar que, confirmándose los datos de los ensayos fase II disponibles, nos hallemos ante fármacos de eficacia superior a los actuales, pero cuya seguridad será necesario ajustar para obtener perfiles adecuados de beneficio/riesgo.
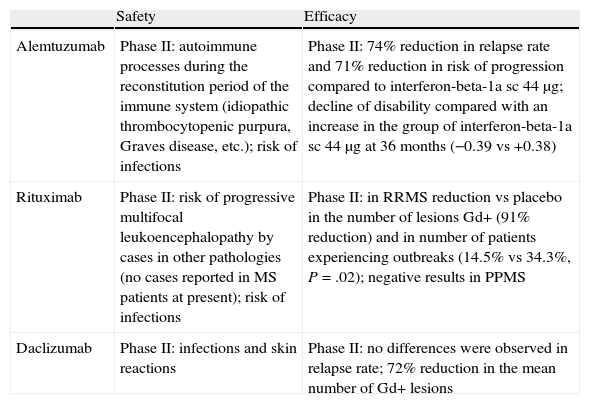
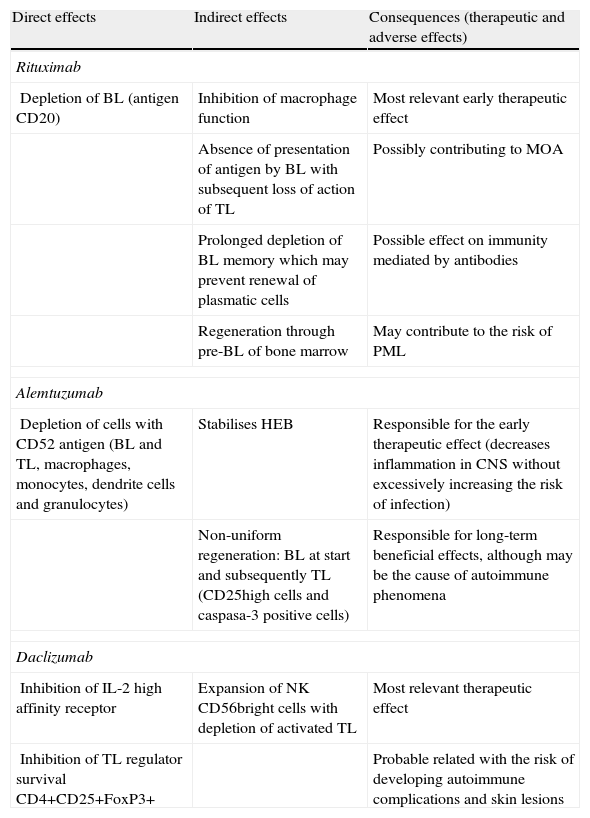
In recent decades, slowly but unstoppably, our therapeutic arsenal in neurology has witnessed the emergence of the first treatments that modify the clinical course of multiple sclerosis. Interferon and glatiramer acetate have recently been followed by natalizumab. It is true that the road ahead is still long, especially to improve comfort during administration and, of course, to improve the efficacy of currently available drugs. The rapid progress we are experiencing in recent years makes it difficult to keep up to date with all ongoing phase II trials, let alone all the molecules that have started preclinical phase I. It is obvious that, along with oral drugs, monoclonal antibodies have been the main focus of this rapid development. We have dedicated this article to a review of the main safety and efficacy results of some monoclonal antibodies that have passed through phase II clinical development: alemtuzumab, rituximab–ocrelizumab and daclizumab. In addition to the references cited, our search for information relied on that available from the following websites: www.clinicaltrials.gov, the EMEA website and the website of the Spanish Medicine Agency. As a summary, Tables 1 and 2 present the fundamental efficacy and safety data on all 3 monoclonal antibodies reviewed, as well as a brief summary of their direct or indirect modes of action.
Main safety and efficacy features of the monoclonal antibodies reviewed in this chapter. Importantly, the difference in study design does not allow a direct comparison of the efficacy data.
| Safety | Efficacy | |
| Alemtuzumab | Phase II: autoimmune processes during the reconstitution period of the immune system (idiopathic thrombocytopenic purpura, Graves disease, etc.); risk of infections | Phase II: 74% reduction in relapse rate and 71% reduction in risk of progression compared to interferon-beta-1a sc 44μg; decline of disability compared with an increase in the group of interferon-beta-1a sc 44μg at 36 months (−0.39 vs +0.38) |
| Rituximab | Phase II: risk of progressive multifocal leukoencephalopathy by cases in other pathologies (no cases reported in MS patients at present); risk of infections | Phase II: in RRMS reduction vs placebo in the number of lesions Gd+ (91% reduction) and in number of patients experiencing outbreaks (14.5% vs 34.3%, P=.02); negative results in PPMS |
| Daclizumab | Phase II: infections and skin reactions | Phase II: no differences were observed in relapse rate; 72% reduction in the mean number of Gd+ lesions |
Gd+: gadolinium-enhanced lesions; MS: multiple sclerosis; PPMS: primary-progressive MS; RRMS: relapsing-remitting MS; sc: subcutaneous.
Modes of action and consequences, both therapeutic and in terms of side effects, of the monoclonal antibodies reviewed.
| Direct effects | Indirect effects | Consequences (therapeutic and adverse effects) |
| Rituximab | ||
| Depletion of BL (antigen CD20) | Inhibition of macrophage function | Most relevant early therapeutic effect |
| Absence of presentation of antigen by BL with subsequent loss of action of TL | Possibly contributing to MOA | |
| Prolonged depletion of BL memory which may prevent renewal of plasmatic cells | Possible effect on immunity mediated by antibodies | |
| Regeneration through pre-BL of bone marrow | May contribute to the risk of PML | |
| Alemtuzumab | ||
| Depletion of cells with CD52 antigen (BL and TL, macrophages, monocytes, dendrite cells and granulocytes) | Stabilises HEB | Responsible for the early therapeutic effect (decreases inflammation in CNS without excessively increasing the risk of infection) |
| Non-uniform regeneration: BL at start and subsequently TL (CD25high cells and caspasa-3 positive cells) | Responsible for long-term beneficial effects, although may be the cause of autoimmune phenomena | |
| Daclizumab | ||
| Inhibition of IL-2 high affinity receptor | Expansion of NK CD56bright cells with depletion of activated TL | Most relevant therapeutic effect |
| Inhibition of TL regulator survival CD4+CD25+FoxP3+ | Probable related with the risk of developing autoimmune complications and skin lesions | |
BL: B-lymphocytes; CNS: central nervous system; IL: interleukin; MOA: mode of action; NK: natural killer; PML: progressive multifocal leukoencephalopathy; TL: T lymphocytes.
Alemtuzumab or campath-1H is a monoclonal antibody humanized against the CD52 antigen, one of the first available humanized antibodies in fact.1 Because this surface receptor is found in most cells in the immune system (T and B lymphocytes, monocytes and eosinophils), treatment with this antibody causes an intense, lasting depletion of immune system cells. It is important to note that CD52 is not yet expressed in the bone marrow progenitors, which do not suffer the effects of the antibody. It should also be stressed that this depletion is not as extreme in the lymphoid organs, which may explain the relatively low rate of infections suffered by patients treated with this antibody. It is possible that neutrophil cell populations and natural killer lymphocytes may intervene in the cell death mechanisms involved, and that the role of the complement is not as important as was previously thought.2 It is already being marketed in Spain under the name MabCampath® for the treatment of B-cell chronic lymphocytic leukaemia in patients in whom chemotherapy treatment in combination with fludarabine is not appropriate (the data sheet can be found at: http://www.ema.europa.eu/humandocs/PDFs/EPAR/mabcampath/emea-combined-h353es.pdf).1,3,4
Clinical evidence available on efficacyThe first published clinical trials with this drug revealed that its use in stages of the disease with little inflammatory component would be a low-yield strategy. Consequently, those responsible for the phase II study design decided to include only patients in the earliest stages of the disease1,5; this is why the CAMMS223 phase II study could only include patients with a disease duration under 4 years.6 The key features of the CAMMS223 trial are: randomized 3-year phase II trial that included 334 patients with an EDSS score less than 3 and a maximum disease duration of 3 years. Patients were not blinded to the medication administered (due to the highly recognisable cytokine release syndrome, it was considered impossible to maintain patients blind as to who would receive this antibody) and received the following in a 1:1:1 ratio: (a) interferon beta-1a in subcutaneous dose of 44μg 3 times per week (Rebif®), and (b) alemtuzumab in 2 different doses (12mg/day or 24mg/day). The program for intravenous administration of alemtuzumab was the following: 5 consecutive days in the first month of the trial, and for 3 consecutive days on months 12 and 24 until completing 3 treatment cycles. The trial was stopped (due to safety reasons) when almost all patients had received doses for month 12, but only 25% had received the dose for month 24. As mentioned, both patients and neurologists were not blinded to the treatment administered; only the evaluating neurologist remained blinded to treatment allocation. Although this must be taken into account when assessing the results of this test, it is true that the two primary objectives (EDSS and the presence of outbreaks) yielded very encouraging results. In the group of alemtuzumab patients (both doses combined), the risk of sustained disability progression was reduced by 71% and the relapse rate by 74%, compared with the Rebif 44® group. Although we have no data on the evolution of gadolinium-enhanced lesions, as no MRIs were performed after administration of gadolinium, the results of brain volume measurements were also remarkable. We observed statistically significant differences in the development of atrophy between alemtuzumab patients and Rebif 44® patients between months 12 and 36 (to avoid the pseudo-atrophy effect occurring in the first months after initiation of anti-inflammatory therapy). The brain volume change was −0.2% in patients receiving Rebif 44® and +0.9% in the alemtuzumab group. This last datum, together with an improvement of disability during the study in the group with alemtuzumab treatment, has led to speculation about a possible neuroprotective and even neuroregenerative effects of this monoclonal antibody.
Safety dataThe encouraging efficacy results of the CAMMS223 trial were unfortunately accompanied by the death of 2 patients in the alemtuzumab group. One patient died from cardiovascular disease; this patient had presented risk factors previously. The second patient developed autoimmune thrombocytopenic purpura (a total of 6 purpura cases were detected in trial patients), which caused a brain haemorrhage. There were other cases of autoimmune pathology, mainly autoimmune thyroid disease (49 cases in patients treated with alemtuzumab), possibly as a consequence of aberrant phenomena during the regeneration process of the immune system (believed to be related to different recovery kinetics of B and T lymphocytes [Table 2]). We also detected an increased risk of infections, especially of the respiratory tract, in patients taking alemtuzumab. Furthermore, 3 patients developed recurrent episodes of herpes after the infusions. A control of the infusion reaction through premedication (corticosteroids, paracetamol and antihistamines) was revealed to be important.
Clinical development programThere are two active phase III clinical trials at the present time: CAREMS-I and CAREMS-II. The former is a 2-year study comparing an alemtuzumab dose of 12mg/day with Rebif 44® (ratio 2:1), with a very similar design to the CAMMS223 trial, although it allows recruitment of patients with a disease duration of up to 5 years. The CAREMS-II trial includes patients who have not responded to previous treatment with immunomodulators, with a maximum disease duration of 10 years. Both studies are underway and have completed the recruitment phase.
ConclusionsAlthough efficacy results are very encouraging, the safety profile of this drug still needs to be secured within acceptable limits. We hope that this can be achieved through the patient control measures to prevent the occurrence of serious adverse effects, especially those related to bleeding diathesis.
RituximabMode of actionRituximab is a chimeric monoclonal antibody (rodent/human) against the CD20 antigen present mainly in B-lymphocytes from the pre-B stage until the stage of activated B-cells. These cells undergo lysis after the binding of this antibody, although it is important to emphasise that this does not happen in the earliest haematopoietic precursors nor in the plasma cells, since they do not express the antigen.3,4 This would allow a more rapid regeneration and would thus maintain some degree of immune defence through plasma cells. The lymphopenia induced by this drug is durable and relatively selective for B-cells. In the phase II HERMES trial, only 30.7% of patients presented normal levels of B-lymphocytes 1 year after the infusion, while there were no changes in the levels of T-lymphocytes in the blood.7 It is noteworthy that, despite this, a decrease was also observed in the levels of T-lymphocytes in cerebrospinal fluid.8 Rituximab is already indicated in Spain, under the commercial name Mabthera®, for the treatment (in some cases) of non-Hodgkin lymphoma, chronic lymphocytic leukaemia and rheumatoid arthritis (the data sheet is available from: http://www.ema.europa.eu/humandocs/PDFs/EPAR/Mabthera/emea-combined-h165es.pdf).
Clinical evidence available on efficacyThe HERMES study investigated the efficacy and safety of rituximab in 104 patients with relapsing-remitting multiple sclerosis through a 1-year phase II trial. Patients were administered 1g of the drug on 2 occasions (day 1 and day 15) or placebo (2:1 ratio). A 91% relative reduction was observed in the primary objective (total number of gadolinium-enhanced lesions at weeks 12, 16, 20 and 24 of the study). Statistically significant differences favouring the treatment group were also observed in the proportion of patients with outbreaks (14.5% vs 34.3% in the placebo group). The OLYMPUS9 study recruited 439 patients with primary-progressive multiple sclerosis in a phase II/III study, which was negative for its primary objective. Statistically significant differences were found to favour only the rituximab group in the accumulation of lesions in T2-weighted sequences, with no such differences being observed in the development of brain atrophy. Post hoc analysis seems to show that the drug could be useful in a subgroup of patients with higher gadolinium enhancement and younger age.
Safety dataSafety results from both the HERMES and the OLYMPUS studies include the presence of infusion reactions, which occurred mainly during the first rounds of drug treatment and then decreased to levels comparable to placebo in subsequent rounds. An increased risk of infections was also observed. In addition, the HERMES study found 1 coronary syndrome and 1 malignant thyroid tumour in the rituximab group. Nevertheless, its use in other indications has reported up to 57 cases of progressive multifocal leukoencephalopathy in patients who had taken rituximab10 (although it this figure might be higher given the non-mandatory reporting of these cases). However, it is noteworthy that many of these patients already suffered from diseases which by themselves predisposed towards the onset of progressive multifocal leukoencephalopathy. Furthermore, the patients had received other immunosuppressive therapies in the vast majority of cases, although 1 of these cases involved a patient with autoimmune haemolytic anaemia who had received only corticosteroids and rituximab.10
Clinical development programAt the present time, clinical development in multiple sclerosis continues through a humanized anti-CD20 compound: ocrelizumab (WA21493 phase II trial), and a human anti-CD20 compound: ofatumumab (GEN414 phase I/II dose finding trial). However, the clinical development of ocrelizumab in rheumatoid arthritis and systemic lupus erythematosus has recently been stopped due to an unacceptable safety profile in these two conditions (http://www.genengnews.com/analysis-and-insight/ocrelizumab-one-size-does-not-fit-all/77899315/).
ConclusionsThe efficacy results obtained in the HERMES phase II study are highly encouraging. However, its safety profile should be defined further in patients with multiple sclerosis, especially with regard to the possibility of developing progressive multifocal leukoencephalopathy symptoms. It is also necessary to obtain more information on the actual risk of infection, which has led to the interruption of the ocrelizumab development program in some pathologies.
DaclizumabMode of actionDaclizumab is a humanized monoclonal antibody against the CD25 surface molecule, which is the alpha chain of the interleukin-2 receptor, a crucial proinflammatory cytokine in the process of T-cell activation. Daclizumab binds selectively to the alpha subunit of the receptor, which is a constituent part of the high- and low-affinity recipients of interleukin-2. It does not form part of the intermediate-affinity receptors (which only consist of beta and gamma subunits), which are therefore not blocked. This antibody was initially tested in multiple sclerosis under the assumption of inhibition of the processes that stimulate autoreactive T-lymphocytes (which do not have intermediate affinity receptors) and which are mediated by interleukin-2.11 Nevertheless, it appears that the beneficial effect in the case of multiple sclerosis is caused by an excess of circulating interleukin-2, which in turn causes a proliferation of CD56bright cells (which act as regulatory cells). These cells do have intermediate-affinity receptors not blocked by daclizumab.12 Moreover, recent works indicate that the increase in autoimmune phenomena, mainly skin problems, observed with this drug could be due to a reduction of regulatory T-cells CD4+CD25+Foxp3+.13 The drug was previously marketed under the name Zenapax®; however, its European authorisation has been cancelled at the request of the pharmaceutical laboratory itself due to commercial reasons not linked to safety alerts (available from: http://www.emea.europa.eu/humandocs/PDFs/EPAR/Zenapax/68376508en.pdf). Its use was approved to prevent acute rejection of kidney transplants.
Clinical evidence available on efficacyThe first open clinical trial showed a 78% reduction in the number of new gadolinium-enhanced lesions. This was an open study including only 10 patients with different forms of multiple sclerosis (relapsing-remitting and secondary-progressive), who did not respond to first-line drugs. In this study, subcutaneous daclizumab was added to interferon-beta.14 A similar trial (although most patients followed monotherapy with daclizumab this time) conducted by independent researchers obtained similar results.15 Recently, the results of the CHOICE16 study have been published. This 6-month study followed a randomized, double-blind design. It recruited 230 patients with relapsing-remitting and secondary-progressive (<10%) multiple sclerosis who had suffered an outbreak in the past year while on stable immunomodulatory therapy. Daclizumab (or placebo) was administered subcutaneously every 15 days in 2 possible doses (1mg/kg and 2mg/kg). The results showed a 72% reduction in the mean number of gadolinium-enhanced lesions for the high dose, without significant results being found for the low dose (1.32 in the group with 2mg/kg vs 3.58 in the group with 1mg/kg vs 4.75 in the group with placebo) and without statistically significant results for the clinical parameters (outbreaks, EDSS, MSFC).
Safety dataThe first open trials found no significant safety issues, although Rose et al.15 did notice 4 patients with skin problems. The safety results of the CHOICE study seemed to confirm these findings and demonstrate the presence of an increased risk of infection, although no increase in the risk of opportunistic infections was found. Two patients in the treated group developed tumours (in situ breast ductal carcinoma and recurrence of pseudomyxoma peritonei).
Clinical development programTwo studies are being conducted at present. One is a phase II study with different doses of daclizumab (150mg and 300mg) administered monthly (DAC HYP) in monotherapy controlled with placebo (205-MS-201), with an extension period (205-MS-202 [SELECT study]). The other is a phase III study with a single dose of daclizumab (150mg) administered monthly (DAC HYP) with an active comparison group treated with interferon beta-1a (DECIDE study).
ConclusionsOngoing monotherapy studies will help us to better understand the efficacy and safety profile of this monoclonal antibody, since at present the high-quality data obtained are mainly from the CHOICE study, with an add-on design.
ConclusionsAlthough there is currently a significant amount of data regarding the efficacy and safety of the three monoclonal antibodies reviewed, it is still too early to decide which position they will occupy within the therapeutic arsenal that will be available shortly for the treatment of multiple sclerosis. If the data available from phase II trials are confirmed, the efficacy parameters would appear to be higher than first-line drugs available at present. Nevertheless, it is crucial to continue accumulating short- and long-term safety data on these drugs. These data will shape the future of the 3 monoclonal antibodies under development for multiple sclerosis reviewed in this chapter. When the time comes for their clinical application, it will also be critical to develop the management and prevention algorithms required to minimise the risk of adverse effects.
Conflict of interestsJaume Sastre-Garriga has served as a consultant or advisory board member for Novartis, TEVA, Biogen, Almirall, Bayer Schering Pharma and Merck-Serono, and has participated as a speaker at events organised by Novartis, Sanofi-Aventis, Biogen, Almirall, Bayer Schering Pharma and Merck-Serono.
Xavier Montalban has served as a consultant or advisory board member for Novartis, TEVA, Biogen, Sanofi-Aventis, Almirall, Bayer Schering Pharma and Merck-Serono and has participated as a speaker at events organised by Novartis, Sanofi-Aventis, Biogen, Almirall, Bayer Schering Pharma and Merck-Serono.
Please cite this article as: Sastre-Garriga J, Montalban X. Anticuerpos monoclonales en desarrollo en esclerosis múltiple. Neurología. 2011;26:556–62.
