



Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease whose aetiology is unknown. It is characterised by upper and lower motor neuron degeneration. Approximately 90% of cases of ALS are sporadic, whereas the other 10% are familial. Regardless of whether the case is familial o sporadic, patients will develop progressive weakness, muscle atrophy with spasticity, and muscle contractures. Life expectancy of these patients is generally 2 to 5 years after diagnosis.
DevelopmentIn vivo models have helped to clarify the aetiology and pathogenesis of ALS, as well as the mechanisms of the disease. However, as these mechanisms are not yet fully understood, experimental models are essential to the continued study of the pathogenesis of ALS, as well as in the search for possible therapeutic targets. Although 90% of cases are sporadic, most of the models used to study ALS pathogenesis are based on genetic mutations associated with the familial form of the disease; the pathogenesis of sporadic ALS remains unknown. Therefore, it would be critical to establish models based on the sporadic form.
ConclusionsThis article reviews the main genetic and sporadic experimental models used in the study of this disease, focusing on those that have been developed using rodents.
La esclerosis lateral amiotrófica (ELA) es una patología neurodegenerativa, progresiva y de etiología desconocida caracterizada por la degeneración de motoneuronas superiores e inferiores. Aproximadamente el 90% de los casos de ELA son esporádicos mientras que el 10% restante se consideran familiares. Independientemente de si son familiares o esporádicas, los pacientes desarrollan una debilidad progresiva, atrofia muscular con espasticidad y contracturas. Por lo general, la esperanza de vida en los pacientes de ELA es de 2 a 5 años.
DesarrolloLos modelos in vivo han ayudado a explicar la etiología y la patogénesis, así como los mecanismos de la esclerosis lateral amiotrófica. Sin embargo, estos mecanismos no están del todo esclarecidos aún, por lo que los modelos experimentales son fundamentales para continuar con el estudio de los mismos, así como para la búsqueda de posibles dianas terapéuticas. A pesar de que el 90% de los casos son esporádicos, la mayoría de los modelos utilizados hasta la actualidad para estudiar la patogénesis están basados en las mutaciones genéticas asociadas a la patología familiar, lo que provoca que la patogénesis de la ELA esporádica no sea aún conocida., Por tanto, sería fundamental el estudio de la enfermedad en modelos basados en la patología esporádica.
ConclusiónEn el presente artículo se han revisado los principales modelos experimentales tanto genéticos como esporádicos utilizados en el estudio de esta enfermedad, enfocándonos en los que se han desarrollado utilizando el roedor como plataforma experimental.
Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative paralytic disease characterised by the degeneration of upper and lower motor neurons, both in the cortex and in the spinal cord.1–5 Prevalence is estimated at 3–5 cases per 100 000 population.6,7 Approximately 90% of cases of ALS are sporadic, whereas 10% are considered familial cases.6–9 Among familial cases, 20% are due to a mutation in the copper–zinc superoxide dismutase-1 antioxidant enzyme (SOD1).7
Whether familial or sporadic, ALS causes progressive weakness and muscular atrophy with spasticity and contractures. Onset of progressive weakness may be distal or proximal, in the upper or lower part of the limbs, eventually affecting all muscles, including those involved in breathing, speech, and swallowing.7,8 Most patients die due to respiratory failure at 2 to 5 years after diagnosis.2,3,6,8
When upper motor neurons are affected initially, patients display muscular rigidity and spasticity. However, initial involvement of lower motor neurons is associated with excessive electric irritability, beginning with spontaneous fasciculations. The main pathological characteristic is death of motor neurons in the motor cortex and spinal cord. In addition to this degeneration of motor neurons, a neuroinflammatory process causes proliferation of astroglia, microglia, and oligodendroglial cells. A common finding, both in sporadic and familial cases, is cytoplasmic aggregation of proteins, with the most common being TDP43, ubiquitin, and, in sporadic cases, SOD1.6
As the mechanisms underlying this disease are not fully understood,10 the development of experimental models is essential to clarifying them and identifying new possible treatments. This article includes an update on the main experimental models used in the study of ALS, which have been reviewed previously by our research group,7 focusing on models developed using rodents.
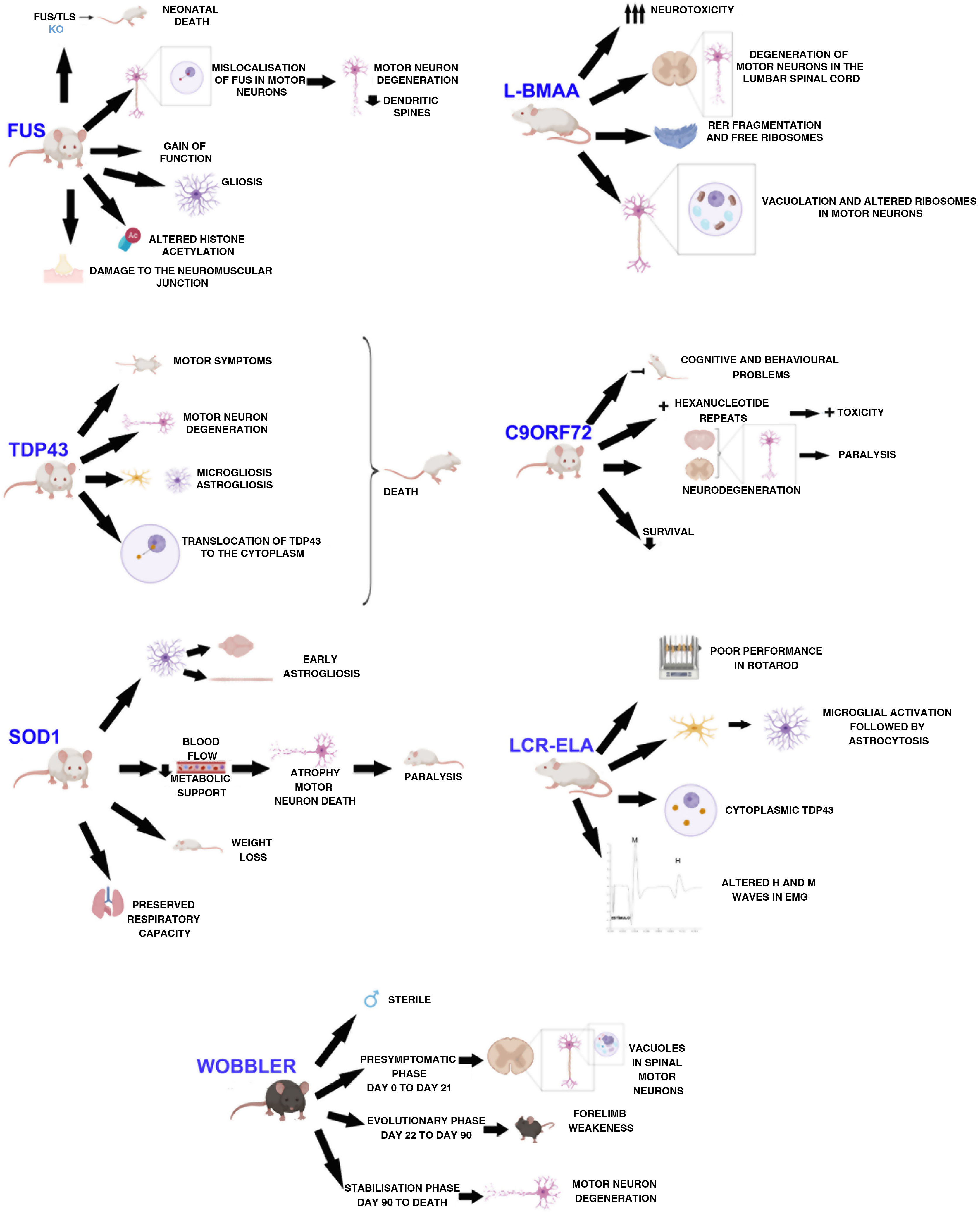
DevelopmentWobbler mouseThis mouse model was described by Falconer in 1956.11 It consists of a spontaneous autosomal recessive mutation in the C57BL/F strain.12,13 This mutation is located on chromosome 11, which is homologous to the 2p13 region in the human genome.12,14,15 The mutation, associated with motor neuron degeneration, is caused by a point mutation in the last exon of the Vps54 gene, causing an amino acid substitution12–15 of leucine with glutamine in the C-terminal domain of the Vsp54 protein.12,15 One of the main characteristics of this model is that males are sterile, despite the normal appearance of their genitals (Fig. 1).16

The clinical course is divided into 3 phases (Fig. 1): an initial presymptomatic phase, followed by an evolutionary phase, and finally a stabilisation phase. In the presymptomatic phase, which comprises the first 3 weeks of postnatal life, mild non-clinical symptoms are observed, with weight, grip force, and righting reflex remaining normal.12,13,16 However, animals present histopathological signs, including vacuolation in spinal motor neurons,16,17 weak staining of Nissl bodies, and enlarged somas.13 In the evolutionary phase, from 3 weeks of age, mice display clinical, morphological, and molecular changes, including muscle atrophy, weight loss, and tremor in the head. The hind limbs are less severely affected, but weakness in the forelimbs and muscles of the neck develop rapidly. After this rapid progression, symptoms stabilise, thus entering the last phase, which extends until the death of the animal. This phase is characterised by the arrest of motor neuron degeneration.12,13,16 In this phase, vacuolation in motor neurons is rarely observed. However, deficiencies in neuromuscular synapses are observed.
This model has been used to test the effectiveness of treatments for motor neuron degeneration. The main disadvantage of this model is that, although the mutation is spontaneous and may be useful to study sporadic cases of ALS, it has not been identified in humans.12 Therefore, it is considered a good model to study motor neuron diseases, rather than ALS specifically.12,13
SOD1In 1993, Rosen et al.18 identified mutations associated with ALS in the SOD1 gene (chromosome 21).19,20 This protein is a cytosolic metalloenzyme composed of 153 amino acids.20 Its physiological function is to catalyse the conversion of superoxide anions to hydrogen peroxide.1,19,21 Mutations in this gene were the first cause of ALS to be described,1,19,20 and account for a total of 20% of cases of familial ALS and 1%–2% of sporadic cases.1
The first animal model carrying mutant human SOD1 was created in 1994, with the SOD1-G93A mutation.8,22 This is the most widely used model in the study of ALS,23,24 although there are more than 10 mutant SOD1 models, such as SOD1-G37R, SOD1-G85R, and SOD1-G86R.25 The different SOD1 models differ in the age of clinical symptom onset, progression, and survival time, according to the specific mutation, number of copies, level of protein expression, and sex.8
The main findings in SOD1 models are early astrogliosis and microgliosis, glutamate-mediated excitotoxicity, impairment of axonal transport, mitochondrial vacuolation, aberrant neurofilament processing, and decreased metabolic support of motor neurons (Fig. 1).1,26,27
The disease was initially believed to be caused by a loss of activity; however, SOD1 knock-out (KO) mice did not develop ALS.8,22
Animals with the SOD1-G93A mutation overexpress the human protein. These animals present clinical and neuropathological signs corresponding to familial ALS, such as paralysis of the limbs and skeletal muscle atrophy.28,29 They also show alterations in dendritic structures of the upper motor neurons, pyramidal neurons of the prefrontal cortex, and the lower motor neurons of the brainstem and spinal cord.23 Muscle denervation precedes the loss of motor neurons and muscle atrophy.29 A longitudinal study by Marcuzzo et al.28 showed that neurodegeneration in this model occurs in the ventral white matter and peripheral nerves from week 8, before extending to the ventral grey matter in week 10. Despite motor neuron death, respiratory capacity is preserved until the end of the disease.30
A recent study in these animals found that muscle contractility precedes the reduction in motor unit connectivity. The authors also showed that muscle atrophy occurs after these processes.31
The reduced blood flow found in SOD1-G93A models and the increased glucose consumption in the spinal cord during the presymptomatic phase suggest that the loss of oxygen and metabolism uncoupling would induce motor neuron death.10
This model has enabled researchers to discover that the progressive decline in corticospinal and bulbospinal projections occurs before clinical symptom onset. Furthermore, presymptomatic and symptomatic SOD1-G93A mice show clear astrogliosis, both in the brain and in the spinal cord. In the last phase of the disease, when motor neurons are already lost, an increase is observed in the number of activated astrocytes in the spinal cord, motor trigeminal nucleus of the brainstem, and primary motor cortex.25
Respiratory capacity is preserved in SOD1-G93A rats despite the loss of phrenic nerve motor neurons, as previously mentioned (Fig. 1). Furthermore, activity of the phrenic nerve decreases by approximately 50%, which suggests a mechanism (plasticity) in the synaptic inputs to compensate for phrenic nerve motor neurons, thus increasing their individual activity.30
Excitotoxicity may manifest due to changes to the intrinsic electrical properties of motor neurons or due to an increase in the properties of excitatory synapses. In the SOD1-G93A model, intrinsic excitability of motor neurons occurs during the early stages, even during the embryonic stage.32
Mice overexpressing SOD1-G93A present no alterations during the first months of life, and start to develop clinical signs from the age of 4 months. They present decreased movement and difficulty moving and extending the limbs, as well as tremor, asymmetrical weakness in the limbs, and progressive weight loss (Fig. 1); electromyographic data revealed spontaneous sharp waves characteristic of atrophy. Histological findings from these mice include vacuoles mainly in the neuropil of the ventral horn, particularly in the dendrites, and a decreased number of motor neurons in the lumbar spinal cord.33
Using SOD1-G37R mice, El Oussimi et al.34 showed that the degeneration of serotonergic neurons is responsible for hyperreflexia in these animals. This hyperreflexia is a compensatory mechanism for motor deficits, thus maintaining motor function after onset of the disease.34 A recent study using these mice has shown that there are differences between sexes in the formation of new axonal branches; despite the increase in these new branches in female mice, these animals also presented greater neuronal loss and denervation of neuromuscular junctions, which may be detrimental to disease progression.35
Although this model develops the majority of the neuropathological findings of ALS,36,37 it is yet to be shown whether these findings may be extrapolated to other forms of familial or sporadic ALS. Specifically, this mutant model has been criticised due to its poor predictive value in testing potential therapies.36
TDP43The TDP43 protein was discovered due to its effects in the transcription of the human immunodeficiency virus.20 It is encoded by the TARDBP gene, located on chromosome 1, and contains a nuclear localisation signal (NLS),20,22,38 2 RNA-binding domains, and a glycine-rich region, in which the majority of pathogenic mutations for ALS have been identified.20 It participates in RNA processing and splicing regulation,20,22,38 and is critical to the functioning and survival of neurons, as neurotoxicity increases if the NLS is altered.38 Therefore, this protein is the main pathological component of cytoplasmic inclusions in ALS and frontotemporal dementia (FTD). Mutations in this protein account for 4% of cases of familial ALS,22 although inclusions are identified in the majority of cases of ALS.22,39
The first transgenic model of TDP43 proteinopathy was created by Wegorzewska et al.40 in 2009. This model expresses the A315 T mutation controlled by a mouse promoter. These animals generate approximately 3 times more mutant protein than endogenous protein, expressing the transgene in the brain and spinal cord. These animals first show motor symptoms at 3 or 4 months of age; by 4 months and a half, they are unable to support their own weight, using their limbs to drag themselves forward (Fig. 1). Post-mortem examinations show degeneration of motor axons and a 20% decrease in the number of motor neurons, with respect to controls. These mice present greater involvement of upper motor neurons than the SOD1 model.40,41
TDP43-induced toxicity depends on the level of transgene expression. Moderate TDP43 expression is associated with an attenuated phenotype, in comparison with animals expressing high levels of TDP43. Both wild-type and mutant TDP43 are neurotoxic when overexpressed.42
Mice overexpressing human TDP43 or an ALS-associated mutation in this gene present accumulation of ubiquitin, TDP43 fragmentation, astrogliosis, microgliosis, axonal degeneration (Fig. 1), neuronal loss, motor dysfunction, and shorter life expectancy.43 Mice overexpressing the M337V mutation present severe motor neuron loss, degeneration of the neuromuscular junction, and premature death (Fig. 1).39
Xu et al.43 used a human TDP-43 transgenic mouse model to determine that levels of the mouse TDP43 protein are significantly decreased with regard to controls, which suggests that the human protein may regulate expression of the mouse protein through downregulation of mouse TARDBP RNA. Despite the decrease in the expression of mouse TDP43, this reduction does not lead to a loss of function, since human TDP43 compensates neuronal loss; therefore, this type of animal would not be an appropriate tool to study the loss of function of mouse TDP43.43
Mice showing inducible expression of the pathogenic A315 T mutation in central nervous system (CNS) neurons show early cortical and hippocampal atrophy, associated with a loss of neurons expressing mutant human TDP43 and reactive astrogliosis. Cytoplasmic location of the protein (Fig. 1), phosphorylated fragments, and elevated ubiquitination in the brain are also observed in these mice. In functional terms, they present progressive memory deficits, hyperactivity, disinhibition, and motor disability, with the latter being associated with expression of mutant TDP43 and its accumulation in alpha motor neurons of the spinal cord.44
TDP43 is a ligand of the oxidation resistance protein 1 (Oxr1). This protein belongs to a family of proteins containing the TLDc domain, which confers protection against oxidative stress. Overexpression of the Oxr1-C isoform reduces cytoplasmic aggregation of TDP43 in M337 V and Q331 K mutant mice, which suggests that Oxr1 potentially alleviates TDP43-associated pathology, such as mitochondrial dysfunction and oxidative stress–induced apoptosis.39
Regarding transgenic TDP43 models, Gendron and Petrucelli42 noted that, as few TDP43 inclusions were observed, aggregation of this protein would not be the initial neurotoxic factor in these animals. However, the mislocalisation and fragmentation observed in several models suggest that these factors may play a role in neurodegeneration.42
FUSFused in sarcoma (FUS) mutations are responsible for 1% of sporadic cases and 4% of familial cases of ALS. FUS is a nuclear protein associated with several stages of gene expression, such as mRNA transcription, splicing, transport, and translation. In neurons, it is found in axons, dendrites, and excitatory synapses. The majority of FUS mutations are missense mutations and occur in the sequence of the C-terminal NLS.45 Since the discovery of the role of FUS in ALS,46 several animal models have been developed using mutations in this protein, including KO mice, and overexpression of both mutant and wild-type FUS.47
FUS/TLS KO mice die immediately after birth, showing infertility and chromosomal instability (Fig. 1).22,48–50 FUS/TLS–deficient neurons show spines with less density and anomalous morphology. This model does not display loss of motor neurons; therefore, pathogenesis seems to be associated with a gain of function.48 Other groups have used the same KO technique to create a model that achieves low expression of the gene with a truncated, non-functional protein. However, in this case, the researchers only studied the model from a reproductive point of view, and not from a neuropathological perspective.49 The neuropathology of the KO model has been studied recently, showing weight loss, a normal number of acetylcholinesterase neurons, and no motor phenotype. The lack of a motor phenotype and neurodegeneration in these mice suggests that FUS depletion alone is insufficient to cause ALS.49
Scekic-Zahirovic et al.45 generated a conditional knock-in mouse model in which the NLS of FUS is deleted. In this model, they observed that FUS was completely mislocalised in the cytoplasm of homozygous mice, which caused degeneration of motor neurons in neonates. Heterozygous mice showed partial FUS-ALS. Thus, these researchers showed that the FUS mutation is associated with degeneration of motor neurons through the mislocalisation of FUS in these cells (Fig. 1), whereas axonal damage and demyelination occur independently of the expression of mutant FUS in motor neurons.45
The overexpression of FUS, whether wild-type or mutant, triggers the degeneration of motor neurons, suggesting that the mutant protein presents toxic gain of function, leading to more aggressive neurodegeneration.45 Overexpression of wild-type human FUS is associated with a more aggressive phenotype in homozygous mice, which develop motor dysfunction leading to paralysis, and show increased cytoplasmic FUS without ubiquitinated inclusions. Loss of motor neurons in the anterior horn of the lumbar spinal cord, damage to the neuromuscular junction (Fig.1), and gliosis are observed.22,50
Devoy et al.47 created a mouse model by introducing the human FUSDelta14 mutation, associated with ALS, at the FUS gene locus. This model synthesises human FUS-ALS, characterised by onset at middle age and progressive degeneration of motor neurons, with a dominant inheritance pattern. Findings were obtained by behavioural analysis of motor performance, muscle physiology to assess innervation and function of hind limb muscles, and pathological analysis of spinal motor neurons.47
Huang et al.46 created a mutant FUS transgenic rat model that develops severe axonopathy of motor neurons, denervation of the skeletal muscles, and substantial loss of cortical and hippocampal neurons. However, transgenic rats with normal FUS showed a deficit in spatial learning and memory and a significant loss of cortical and hippocampal neurons, accompanied by ubiquitin aggregation and glial reaction, thus developing some symptoms of ALS and FTD.46
Several recent studies with animal models have demonstrated aberrant homeostasis of epigenetic marks. One of the altered marks is histone acetylation. Neuron survival is affected when there is a decrease in histone acetylation (Fig. 1). Rossaert et al.51 assessed the effectiveness of a histone deacetylase inhibitor (ACY-738) on the phenotype of a FUS mouse. Their results showed that treatment with this inhibitor restored histone acetylation and metabolic pathways in the spinal cord, which slowed disease progression. This suggests that histone deacetylases may be a possible therapeutic target.51
C9ORF72Hexanucleotide repeat expansions in C9ORF72 are the most frequent genetic cause of ALS (accounting for approximately 35%–45% of cases of familial ALS). The function of C9ORF72 remains unknown, but it has been suggested that it plays a role in protein trafficking.52–54 Expansions occur in non-coding regions.53 Three possible mechanisms may explain the pathogenesis of these expansions: sequestering of RNA-binding proteins, toxicity mediated by dipeptides formed as a result of repeat-associated non-ATG translation, and haploinsufficiency.52,55,56
Using mice with a chronic reduction of C9ORF72 or expressing the human gene with different expansions, researchers have identified a gain of toxicity as the central mechanism of the disease in the CNS of mammals.53 Deletion of both alleles of the gene results in splenomegaly, enlarged cervical lymph nodes, and early death, in addition to cognitive deficits in working memory and increased anxiety (Fig. 1).22,53
When expression of the expansion mutation is driven by human regulatory regions, mice show different phenotypes, including paralysis, anxiety-like behaviour, decreased survival, and generalised neurodegeneration in the brain and spinal cord, with significant degeneration in the cortex and hippocampus57 (Fig. 1).
Koppers et al.52 suggest that the loss of C9ORF72 alone is insufficient to cause ALS; however, they do not rule out the possibility that the loss of function modulates the disease process and conditions its onset, severity, and duration.52
In contrast, other researchers focus on the gain of function, observing that AAV-mediated expression of 66 repeats in the mouse brain is associated with findings of ALS, such as TDP-43 pathology and behavioural deficits. Other studies have also reported ubiquitin inclusions, though not in relevant neuron populations, and animals therefore did not develop the motor phenotype.52
Two groups recently created mouse models expressing dipeptide repeat proteins.56,58 Choi et al.58 created a model in which the brain expresses lower levels of the GR80 dipeptide through the control of an inducible promoter. These mice show the same age-dependent deficits in social behaviour and impaired synaptic function as those observed in ALS, in addition to a mild increase in neuronal loss, microgliosis, and astrogliosis. Furthermore, they present deficits in mitochondrial morphology and function. This impairment may be one of the first pathogenic events manifesting in the disease, as it is observed before behavioural and cellular deficits.58 Hao et al.56 also created a transgenic model, specifically expressing poly-PR (GFP-PR28). Heterozygous mice presented motor deficits, loss of Purkinje cells, and activation of microglia and astrocytes in the cerebellum and spinal cord. Furthermore, synaptic transmission was dysregulated in these mice; all these findings are compatible with ALS. However, homozygous mice died prematurely (Fig. 1), presenting severe motor deficits, possibly due to the high toxicity of this dipeptide.56
Chemical modelBeta-N-methylamino-l-alanine (L-BMAA) is a highly neurotoxic, non-proteinogenic, hydrophilic amino acid produced by cyanobacteria (Fig. 1).36 This amino acid is associated with several neurodegenerative diseases.36,59,60 The first association between l-BMAA and neurodegenerative diseases was established in Guam, with a relationship being observed between this amino acid and the ALS/parkinsonism-dementia complex, although this hypothesis is controversial.59 In patients with ALS, l-BMAA accumulates in the CNS either as a free amino acid or integrated in proteins. Both sporadic and familial cases show these aggregates in the brain, which would suggest that such mechanisms as autophagy are affected.61
Three mechanisms were postulated to explain the neurotoxic effect of this component: direct activation of NMDA receptors, activation of metabotropic glutamate receptors, and induction of oxidative stress.59
Using intraperitoneal injections, De Munck et al.59 analysed the dose to be administered and its effects, in order to establish a correlation between the results obtained and such diseases as ALS, in order to find a model of sporadic ALS. According to their results, doses of 300 mg/kg for 5 consecutive days are ideal for this model. In treated animals, they observed no differences in weight, establishing that general development was not affected. These animals presented neuronal deficits beginning the day after treatment onset, as well as difficulties in motor coordination. Using electron microscopy, the authors observed that motor neurons were affected in the lumbar spinal cord, with abundant endoplasmic reticulum fragmentation and numerous free polyribosomes, in addition to inflammation, vacuolation, and mitochondrial alterations in spinal neurons (Fig. 1)59; therefore, this model presents several neuropathological findings found in human ALS.36
In a subsequent study, the same research group characterise this model, focusing on the possible morphometrical changes and alterations in neurotransmitter levels. The results of that study show that there are several phases of symptom progression in the treated animals. In the first phase (between the first and the third months after treatment), symptoms progress rapidly, possibly due to the excitotoxicity caused in the CNS. Subsequently, there is a second stabilisation stage in which symptoms develop more slowly, mainly due to l-BMAA accumulation in the brain. Using in vivo MRI studies, a progressive loss of muscle volume was observed in the limbs of animals as compared to controls. No change was observed in the total volume of the cerebral cortex. However, the volume of the lateral ventricles was enlarged in treated animals.36 The researchers also studied neurochemical changes in the short term using high-performance liquid chromatography and in the long term with MR spectroscopy. In the short term, they observed high levels of glutamate and taurine in the motor cortex, whereas levels of GABA were low. In the long term, glutamate concentrations were significantly increased compared to controls, and GABA levels were significantly decreased; no differences were observed in taurine levels. These altered levels are associated with excitotoxic mechanisms participating in neurodegeneration.36
Cerebrospinal fluid infusionThe pathogenesis of sporadic ALS is not understood, and the majority of findings are from studies with transgenic models that do not fully replicate sporadic disease.62 Several studies have confirmed the cytotoxic effect of cerebrospinal fluid (CSF) from patients with ALS in cell cultures.63,64 With these results, and considering the proximity between the CSF and the spinal cord, the CSF may be considered as a mechanism for diffusion of the disease.62,63 Some researchers have observed that acute, local injection of ALS-CSF causes changes in the sodium and potassium voltage-gated ion channels.64 Other studies reported that oxidative stress was induced in motor neurons.65 Rats injected with ALS-CSF presented poorer performance in the Rotarod test (Fig. 1) than the baseline levels obtained in control animals; furthermore, the grip force of these animals was severely impaired. The neuronal activity of the motor cortex was also affected.62 A more recent study using intrathecal injection of ALS-CSF reported atrophy of the muscle fibres, decreased the complexity of neuromuscular junctions, ultrastructural damage, and increased oxidative stress, suggesting that skeletal muscle is involved in the pathogenesis of ALS.66 However, local injections do not imitate the temporal course of ALS, and therefore do not reproduce the disease.9 Therefore, our group decided to study the histopathological and functional changes occurring after continuous infusion of ALS-CSF. Osmotic minipumps (Alzet) were filled with cytotoxic CSF from patients with ALS and implanted subcutaneously. The pumps were connected to a brain infusion cannula (Alzet), which was implanted in the right lateral ventricle of the animals. The results of this study show similar cytohistochemical changes in the brain and spinal cord to those found in patients with ALS. We observed microglial activation followed by astrogliosis (Fig. 1), which is found both in patients with ALS and in the SOD1-G93A model. This may support the idea of neuroinflammation as an early event in ALS pathogenesis. We also observed overexpression of S100B, which was correlated with survival. The results also showed presence of TDP43 in the cytosol, which was co-localised with ubiquitin (Fig. 1), as well as proteins associated with Bunina bodies. Levels of metallothionein were also increased, which may indicate a neuroprotective effect in response to infusion of cytotoxic ALS-CSF. These findings may lead to a better understanding of the pathogenesis and progression of ALS.9
ConclusionsIn conclusion, each of the models developed in rodents presents physiological characteristics under the umbrella of ALS, enabling their use in trials and in the development of new drugs. The continuous advance and development of biomedical research and technology has permitted us to continue working in preclinical research based on these animal models. It is essential to understand the limitations of each model, and to recognise that they describe only one specific scenario; therefore, research should continue with the sole aim of finding therapeutic targets for the treatment of ALS.
Conflicts of interestThe authors have no conflicts of interest to declare.
