



La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa, progresiva y de etiología desconocida caracterizada por la degeneración de motoneuronas superiores e inferiores. Aproximadamente el 90% de los casos de ELA son esporádicos, mientras que el 10% restante se consideran familiares. Independientemente de si son familiares o esporádicas, los pacientes desarrollan una debilidad progresiva, atrofia muscular con espasticidad y contracturas. Por lo general, la esperanza de vida en los pacientes de ELA es de 2 a 5 años.
DesarrolloLos modelos in vivo han ayudado a explicar la etiología y la patogénesis, así como los mecanismos de la ELA. Sin embargo, estos mecanismos no están del todo esclarecidos aún, por lo que los modelos experimentales son fundamentales para continuar con el estudio de los mismos, así como para la búsqueda de posibles dianas terapéuticas. A pesar de que el 90% de los casos son esporádicos, la mayoría de los modelos utilizados hasta la actualidad para estudiar la patogénesis están basados en las mutaciones genéticas asociadas a la enfermedad familiar, lo que provoca que la patogénesis de la ELA esporádica no sea aún conocida. Por tanto, sería fundamental el estudio de la enfermedad en modelos basados en la enfermedad esporádica.
ConclusiónEn el presente artículo se han revisado los principales modelos experimentales, tanto genéticos como esporádicos, utilizados en el estudio de esta enfermedad, enfocándonos en los que se han desarrollado utilizando el roedor como plataforma experimental.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease whose aetiology is unknown. It is characterised by upper and lower motor neuron degeneration. Approximately 90% of cases of ALS are sporadic, whereas the other 10% are familial. Regardless of whether the case is familial or sporadic, patients will develop progressive weakness, muscle atrophy with spasticity, and muscle contractures. Life expectancy of these patients is generally 2–5 years after diagnosis.
DevelopmentIn vivo models have helped to clarify the aetiology and pathogenesis of ALS, as well as the mechanisms of the disease. However, as these mechanisms are not yet fully understood, experimental models are essential to the continued study of the pathogenesis of ALS, as well as in the search for possible therapeutic targets. Although 90% of cases are sporadic, most of the models used to study ALS pathogenesis are based on genetic mutations associated with the familial form of the disease; the pathogenesis of sporadic ALS remains unknown. Therefore, it would be critical to establish models based on the sporadic form.
ConclusionThis article reviews the main genetic and sporadic experimental models used in the study of this disease, focusing on those that have been developed using rodents.
La esclerosis lateral amiotrófica (ELA) es un desorden neurodegenerativo, progresivo y paralítico caracterizado por la degeneración de motoneuronas superiores e inferiores tanto corticales como de la médula espinal1-5. La prevalencia de esta enfermedad es de 3-5 casos cada 100.0006,7. Aproximadamente el 90% de los casos de ELA son esporádicos (SALS), mientras que el 10% restante se consideran familiares (FALS)6-9. Dentro de este 10% de casos familiares un 20% son debidos a la mutación de la enzima antioxidante cobre-cinc superóxido dismutasa (SOD1)7.
Independientemente de si son familiares o esporádicas, los pacientes desarrollan una debilidad progresiva, atrofia muscular con espasticidad y contracturas. La debilidad progresiva puede comenzar de manera distal o proximal en la parte superior o inferior de las extremidades hasta alcanzar todos los músculos, incluyendo aquellos relacionados con la respiración, habla y deglución7,8. Los pacientes mueren, la mayoría, por fallo respiratorio, de 2 a 5 años después del diagnóstico2,3,6,8.
En el caso de que se vean afectadas en primer lugar las motoneuronas superiores, se observa rigidez muscular y espasticidad. Por el contrario, si se ven afectadas las motoneuronas inferiores se observa una irritabilidad eléctrica excesiva, liderada por fasciculaciones espontáneas. Las principales características patológicas son la muerte de las motoneuronas de la corteza motora y de la médula espinal. A esta degeneración de las motoneuronas se le une el proceso neuroinflamatorio que provoca una proliferación de la astroglía, la microglía y las células oligodendrogliales. Un hallazgo común, tanto en los casos esporádicos como familiares, es la agregación citoplasmática de proteínas, siendo las más comunes TDP43, ubiquitina y en casos esporádicos de ELA, SOD16.
Debido a que los mecanismos que subyacen a esta enfermedad no están del todo esclarecidos10, es fundamental el desarrollo de modelos experimentales para esclarecer los mecanismos, así como para la búsqueda de posibles tratamientos. En el presente artículo se ha realizado una actualización de los principales modelos experimentales utilizados en el estudio de esta enfermedad previamente revisados en nuestro grupo de investigación7, enfocándonos en los que se han desarrollado utilizando al roedor como plataforma experimental.
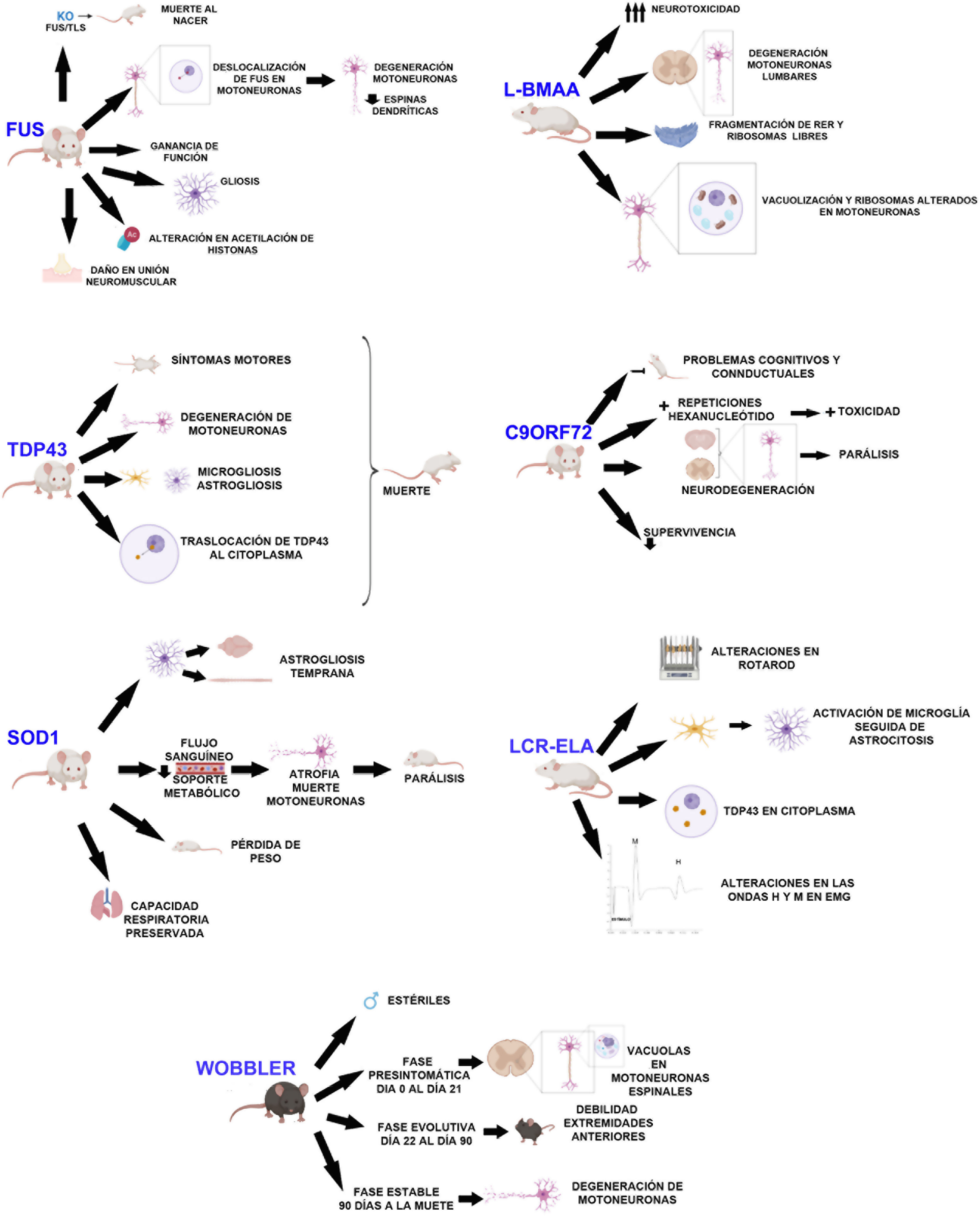
DesarrolloRatón WobblerEste modelo de ratón fue descubierto por primera vez por Falconer en 195611. Consiste en una mutación autosómica recesiva espontánea en la cepa C57BL/F12,13. Dicha mutación se localiza en el cromosoma 11, la cual es homóloga de la región del genoma humano 2p1312,14,15. Está relacionada con la degeneración de las motoneuronas y es causada por una mutación puntual en el último exón del gen Vps54, provocando un cambio de aminoácido12-15 de leucina a glutamina en el dominio C-terminal de la proteína Vps5412,15. Una de las principales características de este modelo es que los machos, a pesar de la normal apariencia de los aparatos genitales, son estériles16 (fig. 1).

Los síntomas que presenta se dividen en 3 fases (fig. 1): una primera fase presintomática, una evolutiva y, por último, una fase estable. En la fase presintomática, cuya duración es desde el nacimiento hasta las 3 semanas, se observan pequeños síntomas no clínicos siendo el peso, la fuerza de agarre y el reflejo de enderezamiento normales12,13,16. Sin embargo, sí se observan rasgos histopatológicos, como por ejemplo la vacuolización en las motoneuronas espinales16,17, marcaje débil de los cuerpos de Nissl y somas aumentados13. Durante la fase evolutiva, que se extiende hasta los 3 meses de edad, se observan cambios clínicos, morfológicos y moleculares, como, por ejemplo, la pérdida muscular y de peso, y el desarrollo de temblor en la cabeza. Las extremidades traseras se ven menos afectadas, sin embargo, la debilidad de las extremidades anteriores y músculos del cuello se desarrolla rápidamente. Tras la rápida progresión observada los síntomas se estabilizan, pasando por tanto a la última fase que se alarga hasta la muerte del animal. Esta fase está caracterizada por una detención de la degeneración de las motoneuronas12,13,16. En esta fase es extraño observar motoneuronas vacuolizadas. Sin embargo, se observan deficiencias en las sinapsis neuromusculares16.
Este modelo se ha utilizado para testar la eficacia de tratamientos en el curso de la degeneración de motoneuronas16. La principal desventaja de este modelo es que, a pesar de que es una mutación espontánea y podría servir para estudiar los casos esporádicos de la enfermedad, esta no se ha encontrado en ningún caso humano12. Es por ello que se considera un buen modelo para las enfermedades de motoneurona en vez de para la ELA12,13.
SOD1Rosen et al. descubrieron en 1993 las mutaciones en el gen SOD1 (cromosoma 21) que estaban relacionadas con la ELA18-20. Esta proteína es una metaloenzima de 153 aminoácidos que se expresa en el citosol20. Su función fisiológica es catalizar la conversión de los aniones superóxidos a peróxido de hidrógeno1,19,21. Las mutaciones en este gen fueron la primera causa genética descrita para la ELA1,19,20 y representan un total del 20% de la ELA familiar y un 1-2% de los casos esporádicos1.
El primer modelo animal que portaba una mutación de la SOD1 humana fue creado en 1994, siendo esta la G93A8,22. Este modelo es el más utilizado para el estudio de la ELA23,24, a pesar de que existen más de 10 modelos mutantes de SOD1, como por ejemplo SOD1-G37R, SOD1-G85R o SOD1-G86R25. Los diferentes modelos de SOD1 presentan variabilidad en cuanto a la edad de inicio de los síntomas clínicos, la progresión y la supervivencia, dependiendo de la mutación específica del gen, el número de copias, los niveles de expresión de la proteína o el género8.
Los principales hallazgos en los modelos SOD1 son un inicio temprano de la astrogliosis y microgliosis, excitotoxicidad mediada por glutamato, déficits en el transporte axonal, vacuolización mitocondrial, procesamiento aberrante de los neurofilamentos y soporte metabólico reducido en las motoneuronas1,26,27 (fig. 1).
En un primer momento se pensó que la enfermedad era causada por una pérdida de la actividad, sin embargo, los ratones knock-out (KO) para SOD1 no desarrollaban ELA8,22.
Los animales SOD1-G93A sobreexpresan la mutación humana SOD1G93A. Este animal presenta signos clínicos y neuropatológicos que correlacionan con FALS, como son parálisis de las extremidades con atrofia de los músculos esqueléticos28,29. Además, muestra alteraciones en las estructuras dendríticas de las motoneuronas superiores, neuronas piramidales del córtex prefrontal y en las motoneuronas inferiores del tronco del encéfalo y la médula espinal23. La denervación muscular precede a la pérdida de las motoneuronas y a la atrofia muscular29. En un estudio longitudinal, Marcuzzo et al. encontraron que la neurodegeneración en este modelo sucede desde la semana 8 en la materia blanca ventral y en los nervios periféricos antes de extenderse a la materia gris ventral en la semana 1028. A pesar de la muerte de las motoneuronas, en estos ratones la capacidad respiratoria se preserva hasta el final de la enfermedad30.
En una reciente investigación con estos animales se encontró que la contractilidad de los músculos precedía a la reducción de la conectividad en la unidad motora. Además, demostraron que la atrofia muscular ocurre de forma posterior a estos procesos31.
La reducción del flujo sanguíneo encontrada en los mutantes SOD1-G93A y el aumento del uso de la glucosa en la médula espinal en la fase presintomática sugieren que la pérdida de oxígeno y el desacoplamiento del metabolismo induciría la muerte de las motoneuronas10.
Este modelo ha permitido descubrir que el declive progresivo de las proyecciones corticoespinales y bulboespinales sucede anteriormente al inicio de los síntomas clínicos. Por otro lado, ratones SOD1-G93A presintomáticos y sintomáticos muestran una evidente astrogliosis, tanto en el cerebro como en la médula espinal. En el último estadio de la enfermedad, cuando las motoneuronas ya se han perdido, el número de astrocitos activados está aumentado en la médula espinal, el núcleo motor trigeminal del tronco del encéfalo y en la corteza motora primaria25.
A pesar de la pérdida de las motoneuronas frénicas, las ratas SOD1-G93A, como hemos comentado anteriormente, mantienen la capacidad respiratoria (fig. 1). Además, la actividad del nervio frénico se reduce aproximadamente un 50%, lo que sugiere un mecanismo (plasticidad) compensatorio en los inputs sinápticos para suplir a las motoneuronas frénicas, amplificando así su actividad individual30.
La excitotoxicidad puede aparecer debido a cambios en las propiedades eléctricas intrínsecas de las motoneuronas o debido a un incremento de las propiedades de las sinapsis excitatorias. En el modelo SOD1-G93A la excitabilidad intrínseca de las motoneuronas se produce en los primeros estadios incluso en el estado embrionario32.
Los ratones que sobreexpresan SOD1-G37R no presentan alteraciones durante los primeros meses de vida, sin embargo, comenzaban a desarrollar signos clínicos a partir de los 4 meses de edad. Presentaban disminución del movimiento, dificultad para mover y extender las extremidades, así como temblores, debilidad asimétrica en las extremidades, pérdida de peso progresiva (fig. 1) y los datos electromiográficos presentaban ondas agudas espontáneas características de la atrofia. Los datos histológicos de estos ratones muestran vacuolas principalmente en el neuropilo del asta ventral, particularmente en las dendritas y reducción de las motoneuronas en la zona lumbar de la médula ósea33.
Oussini et al. demostraron, utilizando ratones SOD1 (G37R), que la degeneración de las neuronas serotoninérgicas es la responsable de la hiperreflexia en estos animales. Esta hiperreflexia es un mecanismo compensatorio a los déficits motores, permitiendo el mantenimiento de la función motora tras el inicio de la enfermedad34. En un estudio reciente utilizando estos ratones se ha visto que existen diferencias entre sexos en la formación de nuevas ramas axonales observando que, a pesar de que estas nuevas ramas estaban incrementadas en los ratones hembra, también presentaban mayor pérdida neuronal y denervación de las uniones neuromusculares, lo que podría ir en detrimento de la progresión de la enfermedad35.
A pesar de que este modelo desarrolla la mayoría de los hallazgos neuropatológicos de ELA36,37, queda por demostrar aún si estos hallazgos pueden extrapolarse a otras formas de ELA familiar o esporádica. En concreto, este mutante ha sido criticado debido a su poco valor predictivo para testar potenciales terapias36.
TDP43La proteína TDP43 se descubrió debido a sus efectos en la transcripción del virus de la inmunodeficiencia humana20. Está codificada por el gen TARDBP en el cromosoma 1. Contiene una señal de localización nuclear (NLS)20,22,38, 2dominios de unión a RNA y una región rica en glicina, región en la cual se han identificado la mayoría de las mutaciones patogénicas para ELA20. Desempeña un papel en el procesamiento del ARN y en la regulación del splicing20,22,38, además de ser crítica para la función y la supervivencia de las neuronas debido a que, si se altera la NLS, aumenta la neurotoxicidad38. Esto provoca que esta proteína sea el mayor componente patológico de las inclusiones citoplasmáticas en la ELA y la demencia frontotemporal (DFT). Las mutaciones de esta proteína representan al 4% de las FALS22, pero las inclusiones se encuentran en la mayoría de los casos de ELA22,39.
El primer modelo transgénico de proteinopatía TDP43 fue creado por Baloh et al. en 2009. Este modelo expresa la mutación A315T bajo el control de un promotor de ratón. Estos animales generan aproximadamente 3 veces más proteína mutada que la endógena, expresándose el transgén en el cerebro y la médula espinal. En primer lugar, estos animales muestran síntomas motores a los 3 o 4 meses de edad, y a los 4 meses y medio ya no son capaces de soportar su propio peso utilizando sus extremidades para deslizarse (fig. 1). Las necropsias de estos animales muestran degeneración de los axones motores y también se observa que presentan una disminución del 20% en el número de motoneuronas respecto a los controles. Estos ratones presentan una mayor afectación de las motoneuronas superiores que el modelo SOD140,41.
La toxicidad inducida por TDP43 es dependiente de los niveles de expresión del transgén. Con una expresión intermedia de TDP43 se observa un fenotipo atenuado en comparación con los animales que expresan altos niveles de TDP43. Tanto TDP43 wild type como la mutante son neurotóxicas si se sobreexpresan42.
Los ratones que sobreexpresan la TDP43 humana o una mutación de este gen asociada a la ELA presentan acumulación de ubiquitina, fragmentación de la TDP43, astrogliosis, microgliosis, degeneración axonal (fig. 1), pérdida neuronal, discapacidad motora y esperanza de vida reducida43. Los ratones que sobreexpresan la mutación M337V presentan pérdida severa de motoneuronas, degeneración de la unión neuromuscular y muerte prematura39 (fig. 1).
Xu Ya-Fei et al., utilizando ratones transgénicos para la TDP43 humana, determinan que la proteína TDP43 de ratón está significativamente disminuida respecto a los controles, lo que sugiere que la proteína humana podría regular la expresión de la de ratón a través de la baja regulación del ARN Tardbp. Pero a pesar de que la expresión de la TDP43 de ratón se reduce, esta reducción no lidera la pérdida de función ya que la TDP43 humana compensa la pérdida funcional, por tanto, este tipo de animales no serían una buena herramienta para estudiar la pérdida de la función de mTDP4343.
Los ratones que expresan de forma inducible la mutación patogénica A315T en las neuronas del sistema nervioso central (SNC) muestran atrofia cortical e hipocampal temprana, asociada a la pérdida de las neuronas que expresan la TDP43 humana mutada, y astrogliosis reactiva. También observaron localización citoplasmática de la proteína (fig. 1), fragmentos fosforilados, así como una elevada ubiquitinización en el cerebro de estos ratones. De forma funcional, estos ratones presentan déficits de memoria progresivos, hiperactividad, desinhibición y discapacidad motora, siendo esta última asociada a la expresión de la TDP43 mutada y a su acúmulo en las motoneuronas alfa en la médula espinal44.
TDP43 es un ligando de Oxr1 (proteína de resistencia a la oxidación 1). Esta proteína pertenece la familia de proteínas que contienen el dominio TLDc, el cual confiere la protección frente al estrés oxidativo. La sobreexpresión de la isoforma Oxr1-C reduce la agregación citoplasmática de la TDP43 en los mutantes M337V y Q331K, lo cual sugiere que Oxr1 posee el potencial de reducir la enfermedad asociada a TDP43 como puede ser la disfunción mitocondrial y la apoptosis inducida por el estrés oxidativo39.
Teniendo en cuenta los animales transgénicos para TDP43, Gendron y Petrucelli consideran que, al observar pocas inclusiones de TDP43, la agregación de TDP43 no sería el factor neurotóxico inicial en estos animales. Sin embargo, la deslocalización y la fragmentación observada en varios modelos sugiere que estas pueden desempeñar un papel en la neurodegeneración42.
FUSLas mutaciones de Fused in Sarcoma (FUS) representan el 1% de la enfermedad esporádica y el 4% de la familiar. FUS es una proteína nuclear relacionada con múltiples pasos de la expresión génica, como la transcripción del mRNA, el splicing, el transporte y la traducción. En las neuronas se encuentra en axones, dendritas y sinapsis excitatorias. La mayoría de las mutaciones de FUS son mutaciones con cambio de sentido y se localizan en la secuencia del dominio C-terminal de la localización nuclear (NLS)45. Tras el descubrimiento de la implicación de FUS en la enfermedad de la ELA46 se han desarrollado varios modelos animales utilizando mutaciones de esta proteína, incluyendo knockouts, y sobreexpresión tanto del FUS mutante como del wild-type47.
Los ratones KO FUS/TLS mueren inmediatamente tras el nacimiento mostrando esterilidad e inestabilidad cromosómica22,48-50 (fig. 1). Las neuronas deficientes de FUS/TLS muestran unas espinas con menos densidad y morfología anómala. En este modelo no se produce pérdida de motoneuronas, por lo que la patogénesis estaría relacionada con una ganancia de función48. Otros grupos han usado la misma técnica KO, pero han generado un modelo en el que obtienen una expresión baja del gen con una proteína truncada y no funcional. Sin embargo, en este caso solo se estudió el modelo desde un punto de vista del sistema reproductivo y no desde la neuropatología49. Recientemente, se ha estudiado la neuropatología del modelo KO observándose pérdida de peso, pero no fenotipo motor y siendo el número de las neuronas acetilcolinesterasa positivas normal. La falta del fenotipo motor y la neurodegeneración en estos ratones sugiere que la disminución solo de FUS es insuficiente para causar los síntomas de ELA49.
Scekic-Zahirovic et al. generaron un modelo de ratón condicional knock-in en el cual se eliminaron tanto NLS como FUS. Con este modelo vieron que FUS estaba completamente deslocalizado en el citoplasma de los ratones homocigotos, lo que provocaba la degeneración de las motoneuronas en neonatos. Los ratones heterocigotos muestran parcialmente la enfermedad FUS-ELA. Con ello demostraron que la mutación en FUS está asociada a la degeneración de motoneuronas a través de la deslocalización de FUS en estas células (fig. 1), mientras que el daño axonal y la desmielinización ocurre independientemente de la expresión del mutante FUS en la motoneuronas45.
La sobreexpresión de FUS, tanto wild type como mutante, desencadena la degeneración de las motoneuronas, sugiriendo que la proteína mutante tiene una ganancia de función tóxica que lidera la neurodegeneración más agresiva45. La sobreexpresión de hFUSwt muestra un fenotipo más agresivo en los homocigotos, que desarrollan discapacidad motora, la cual desemboca en parálisis, y muestran un aumento de la FUS citoplasmática pero no son inclusiones ubiquitinadas. Se observa pérdida de motoneuronas en el asta anterior de la médula lumbar, daño en la unión neuromuscular (fig. 1) y gliosis22,50.
Devoy et al. han generado un modelo murino que consiste en introducir en el locus del gen FUS de ratón la mutación FUSDelta14 humana, la cual se relaciona con la ELA. Este modelo sintetiza la enfermedad FUS-ELA humana definida por un inicio con mediana edad y una progresiva degeneración de las motoneuronas con una herencia dominante. Los resultados se obtuvieron del análisis del comportamiento motor, fisiología muscular para evaluar la inervación y la función de los músculos de las extremidades y análisis patológico de las motoneuronas espinales47.
En ratas, Huang et al. han generado un modelo transgénico de FUS mutante, el cual desarrolla axonopatía severa de las motoneuronas, denervación de los músculos esqueléticos y pérdida sustancial de las neuronas corticales e hipocampales. Sin embargo, ratas transgénicas con la FUS normal muestran déficits en la memoria y aprendizaje espacial, pérdida de neuronas corticales e hipocampales, acompañada de agregación de ubiquitina y reacción glial, desarrollando así algunos de los síntomas de la ELA y la DFT46.
Recientemente, se ha encontrado en diversos modelos animales que existe una homeostasis aberrante de las marcas epigenéticas. Una de estas marcas que están alteradas es la acetilación de las histonas. La supervivencia neuronal se ve afectada si existe un descenso en la acetilación de histonas (fig. 1). Rossaert et al. evaluaron la eficacia de un inhibidor (ACY-738) de deacetilasas sobre el fenotipo de un ratón FUS. Encontraron que el tratamiento con este inhibidor reestablecía la acetilación de histonas además de las vías metabólicas en la médula espinal, lo cual provocaba que la progresión de la enfermedad fuera menor. Esto sugeriría que las histonas deacetilasas pueden ser una posible diana terapéutica51.
C9ORF72Las expansiones del hexanucleótido en C9ORF72 son la causa genética más frecuente de la ELA (aproximadamente representa al 35-45% de las FALS). La función de C9ORF72 es desconocida, pero se ha sugerido que tendría un papel en el tráfico de proteínas52-54. Las expansiones ocurren en la parte no codificante del gen53. Se han descrito 3 mecanismos posibles para la patogenicidad de estas expansiones: secuestro de las proteínas de unión al RNA, toxicidad mediada por dipéptidos formados como resultado de la traducción no ATG asociada a la repetición y la haploinsuficiencia52,55,56.
Utilizando ratones con una reducción crónica de C9ORF72 o expresando el gen humano con diferentes expansiones, se ha identificado una ganancia de toxicidad como mecanismo central de la enfermedad en el sistema nervioso central de los mamíferos53. Eliminando los 2alelos del gen, se produce en los ratones esplenomegalia, aumento de los nódulos linfáticos cervicales y muerte prematura, además de déficits cognitivos en memoria de trabajo y evaluación de la ansiedad22,53 (fig. 1).
Los ratones donde la expansión de la mutación está dirigida por las regiones reguladores humanas muestran diferentes fenotipos incluyendo parálisis, comportamiento similar a la ansiedad, descenso en la supervivencia, la neurodegeneración generalizada en el cerebro y la médula espinal, siendo significativo en la corteza y el hipocampo57 (fig. 1).
Koppers et al. sugieren que la pérdida de C9ORF72 por sí misma es insuficiente para causar la ELA, sin embargo, no descartan la posibilidad de que la pérdida de función module el proceso de la enfermedad e influya en el inicio, la severidad y la duración52.
En contraste, otros investigadores se basan en la ganancia de función; utilizando ratones en los que se inducen 66 expansiones mediante AAV, se observa que los cerebros de estos ratones muestran los hallazgos de la ELA como con la enfermedad TDP43 o los déficits de comportamiento. Otros estudios han encontrado también inclusiones ubiquitinadas, pero no en las poblaciones neuronales relevantes y, por lo tanto, sin desarrollar el fenotipo motor52.
Recientemente 2grupos han generado modelos de ratón que expresaban proteínas de repetición de dipéptidos56,58. Choi et al. generaron un modelo en el cual consiguen que los cerebros de estos ratones expresen menores niveles del dipéptido GR80 mediante el control de un promotor inducible. Estos ratones muestran de manera asociada a la edad los mismos déficits de comportamiento social y deterioro de la función sináptica que se observa en la ELA, además de un ligero aumento de la muerte neuronal, microgliosis y astrogliosis. Por otro lado, presentan un déficit en la morfología y en la función de las mitocondrias. Este daño podría ser uno de los primeros eventos patogénicos que suceden en la enfermedad, ya que se observa previamente a los déficits de comportamiento y celulares58. Hao et al. también generaron un modelo transgénico, en el cual los animales expresaban específicamente poly-PR (GFP-PR28). Los ratones heterocigotos presentaban déficits motores, pérdida de células de Purkinje, activación de la microglía y de los astrocitos en el cerebelo además de en la médula espinal. Además, la transmisión sináptica estaba desregulada en estos ratones, todo ello correlaciona con la enfermedad de la ELA. Sin embargo, los ratones homocigotos morían prematuramente (fig. 1) presentado altos déficits motores, posiblemente debido a la alta toxicidad de este dipéptido56.
Modelo químicoBeta-N-metilamino-L-alanina (L-BMAA) es un aminoácido no proteico e hidrofílico producido por cianobacterias. Es altamente neurotóxico36 (fig. 1). Este aminoácido está relacionado con diferentes enfermedades neurodegenerativas36,59,60. La primera relación que se estableció entre L-BMAA y las enfermedades neurodegenerativas fue en la isla de Guam, en la que se vio una relación entre este aminoácido y el síndrome ELA-párkinson ligado a demencias, aunque esta hipótesis es controvertida59. En los pacientes de ELA, L-BMAA se acumula en el SNC como un aminoácido libre o integrado en proteínas. Tanto en la enfermedad esporádica como en la familiar se observan estos agregados en el cerebro, lo que nos indicaría que se ven afectados mecanismos como el de la autofagia61.
Se postularon 3mecanismos por los cuales este compuesto provoca su efecto neurotóxico: la activación directa de los receptores NMDA, la activación de los receptores de glutamato metabotrópicos y la inducción del estrés oxidativo59.
De Munck et al., mediante inyecciones intraperitoneales, realizaron un análisis de las dosis que deben administrar y sus efectos, para así realizar una correlación entre los resultados obtenidos y enfermedades como la ELA, con el fin de buscar un modelo de ELA esporádica. Según sus resultados, la dosis 300mg/kg durante 5 días consecutivos es la dosis idónea para este modelo. En los animales tratados observaron que no existían diferencias en cuanto al peso, por lo que el desarrollo general no se veía afectado. Estos animales presentan déficits neuronales desde el día siguiente al inicio del tratamiento, así como dificultades en la coordinación motora. Mediante microscopia electrónica observaron que las motoneuronas se veían afectadas a nivel de médula espinal lumbar, fragmentación de retículo endoplasmático y polirribosomas libres, además las neuronas espinales presentan inflamación, vacuolización y mitocondrias alteradas59 (fig. 1), por lo que este modelo presenta varios de los hallazgos neuropatológicos encontrados en la ELA humana36.
En un estudio posterior de este mismo grupo, realizaron la caracterización de este modelo centrándose en los posibles cambios morfométricos y de los niveles de neurotransmisores. Los resultados de dicho estudio muestran que existen varias fases en la evolución de los síntomas de los animales tratados. Una primera fase en la que se produce una rápida evolución de los síntomas que ocurre entre el primer y tercer mes tras el tratamiento, posiblemente debido a la excitotoxicidad causada en el SNC. Posteriormente, una segunda fase de estabilización en la cual los síntomas se desarrollan más lentamente, debido principalmente a los reservorios de L-BMAA del cerebro. Mediante resonancias magnéticas in vivo, se observó una pérdida progresiva del volumen muscular de las extremidades en los animales tratados en comparación con los controles. No se observó cambio en el volumen total de la corteza cerebral. Sin embargo, el volumen de los ventrículos laterales se ve agrandado en los animales tratados36. Por otro lado, también estudiaron los cambios neuroquímicos a corto plazo mediante HPLC y a largo plazo mediante espectroscopia de resonancia magnética. A corto plazo, observaron niveles elevados de glutamato y taurina en la corteza motora, mientras que los niveles de GABA se encontraban disminuidos. A largo plazo, las concentraciones de glutamato estaban significativamente aumentadas respecto a los controles, los niveles de GABA estaban significativamente disminuidos, pero en los niveles de taurina no se observaron diferencias. Estos niveles alterados tienen relación con los mecanismos excitotóxicos que están envueltos en la neurodegeneración36.
Infusión líquido cefalorraquídeoLa patogénesis de la ELA esporádica no es conocida y la mayoría de los hallazgos se han realizado utilizando modelos transgénicos que no replican totalmente la enfermedad esporádica62. Existen diferentes estudios que confirman el efecto citotóxico del líquido cefalorraquídeo (LCR) de pacientes de ELA en cultivo celular63,64. Con estos resultados y teniendo en cuenta la cercanía del LCR a la médula espinal, podría plantearse que el LCR fuera un mecanismo de difusión de la enfermedad62,63. Algunos investigadores han observado que la inyección aguda y puntual de LCR-ELA provoca cambios en canales iónicos dependientes de voltaje de sodio y potasio64. Otros estudios indicaron que se induce estrés oxidativo en las motoneuronas65. Ratas tratadas con LCR-ELA muestran que los parámetros obtenidos en la prueba del Rotarod (fig. 1) fueron menores que los niveles basales tomados con animales controles, además de que la fuerza de agarre de estos animales se vio seriamente afectada. También afectó a la actividad neuronal de la corteza motora62. Recientemente, en un estudio donde inyectaban intratecalmente LCR-ELA, observaron atrofia en las fibras musculares, una disminución de la complejidad de las uniones neuromusculares, daño ultraestructural y aumento del estrés oxidativo, lo cual sugeriría que el músculo esquelético está involucrado en la patogénesis de la ELA66. Sin embargo, al ser inyecciones puntuales no imitan el curso temporal de la ELA y no la reproducen9. Es por ello que nuestro grupo se planteó el estudio de los cambios histopatológicos y funcionales tras la infusión continua de LCR-ELA. Utilizando LCR citotóxico de pacientes de ELA, se rellenaron unas minibombas osmóticas (Alzet) las cuales se implantaron vía subcutánea, a las que se adjuntó una cánula de infusión de cerebro (Alzet) que se implantó en el ventrículo lateral derecho de los animales. Los resultados de este estudio muestran cambios citohistoquímicos en cerebro y médula espinal que se asemejan a los encontrados en los pacientes de ELA. Se observó una activación microglial seguida de una astrogliosis (fig. 1), lo cual se observa tanto en pacientes con ELA como en el modelo SOD1-G93A. Dicho evento puede apoyar la idea de que la neuroinflamación es un proceso temprano en la patogénesis de la ELA. También se obtuvo una sobreexpresión de S100B, la cual correlaciona con la supervivencia en la enfermedad. Los resultados mostraron también presencia citosólica de TDP43 colocalizada con ubiquitina (fig. 1), así como proteínas relacionadas con los cuerpos de Bunina. También se incrementaron los niveles de metalotioneína que podría indicar una reacción neuroprotectora en respuesta a la infusión de LCR-ELA citotóxico. Estos hallazgos pueden mejorar la comprensión de la patogénesis y evolución de la ELA9.
ConclusionesEn conclusión, cada uno de los modelos desarrollados en roedores ofrece características fisiopatológicas dentro del mosaico de la ELA, permitiendo su uso en el ensayo y desarrollo de nuevos fármacos. El avance y el desarrollo continuo de la investigación biomédica y tecnológica ha permitido mantener vigentes las investigaciones preclínicas basadas en estos modelos experimentales. Es necesario entender las limitaciones que posee cada modelo y que solo pueden describir un escenario concreto de la patología, por lo que las investigaciones deberían continuar con el único fin de encontrar dianas terapéuticas para el tratamiento de la ELA.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

