



This paper highlights the relationship of inflammation and oxidative stress as damage mechanisms of Multiple Sclerosis (MS), considered an inflammatory and autoimmune disease.
DevelopmentThe oxidative stress concept has been defined by an imbalance between oxidants and antioxidants in favor of the oxidants. There is necessary to do physiological functions, like the respiration chain, but in certain conditions, the production of reactive species overpassed the antioxidant systems, which could cause tissue damage. On the other hand, it is well established that inflammation is a complex reaction in the vascularized connective tissue in response to diverse stimuli. However, an unregulated prolonged inflammatory process also can induce tissue damage.
ConclusionBoth inflammation and oxidative stress are interrelated since one could promote the other, leading to a toxic feedback system, which contributes to the inflammatory and demyelination process in MS.
Este trabajo destaca la relación de la inflamación y el estrés oxidativo como mecanismos de daño de la esclerosis múltiple, considerada enfermedad inflamatoria y autoinmune.
DesarrolloEl concepto de estrés oxidativo se ha definido por un desequilibrio entre oxidantes y antioxidantes a favor de los oxidantes. Es necesario para realizar funciones fisiológicas, como la cadena respiratoria, pero en ciertas condiciones la producción de especies reactivas sobrepasaba los sistemas antioxidantes, lo que podría causar daño tisular. Por otro lado, está establecido que la inflamación es una reacción compleja en el tejido conectivo vascularizado en respuesta a diversos estímulos, pero un proceso inflamatorio prolongado no regulado también puede inducir daño tisular.
ConclusiónTanto la inflamación como el estrés oxidativo están interrelacionados entre sí, ya que uno de ellos podría promover al otro, dando lugar a un sistema de retroalimentación tóxico, que contribuye al desarrollo del proceso inflamatorio y desmielinizante en la esclerosis múltiple.
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS), which gives rise to focal lesions in the gray and white matter and diffuses neurodegeneration in the entire brain.1,2 MS has an unknown etiology, characterized by demyelination and variable degrees of axonal loss. In the early phase of the disease, inflammatory lymphocyte, macrophage, and activated microglia infiltrates, followed by excessive inflammatory mediators production, lead to demyelination and axonal conduction block, leading to neurological disability. MS manifests as CNS plaques and areas of focal demyelination within the white matter of the brain, spinal cord, and optic nerve. MS plaques are associated with blood vessels and periventricular regions with inflammatory infiltrate containing activated macrophages, microglia, T cells, B cells, and plasmatic cells.3,4
The gradual accumulation of disability characteristic of the disease result from a different set of mechanisms such as (a) B-cell dysregulation, (b) CD8+ T cells causing demyelination or axonal/neuronal damage, (c) microglial cell activation associated with neuritic transection found in cortical demyelinating lesions, and (d) oxidative stress through generation of oxygen and nitrogen reactive species and mitochondrial damage. These different mechanisms are not mutually exclusive and could act in combination.5 In this review, we focus on the mechanisms involved in the development of stress oxidative and inflammatory processes in the pathophysiology of MS.
Oxidative stress in MSThe concept of oxidative stress was introduced for research in redox biology and medicine in 19856; it has been defined by an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control molecular damage.7 Increased oxidative stress can induce damage to the cellular structure and potentially destroy tissues; it represents one of the most common denominators of toxicity. All chemical, physical, and microbial agents can lead to oxidative stress in tissues and cells.8,9 However, oxidative reactions are involved in many fundamental life processes, such as cell respiration, lipid synthesis, metal metabolism, lysosomes, phagocytosis, and xenobiotic biotransformation of organic compounds.9
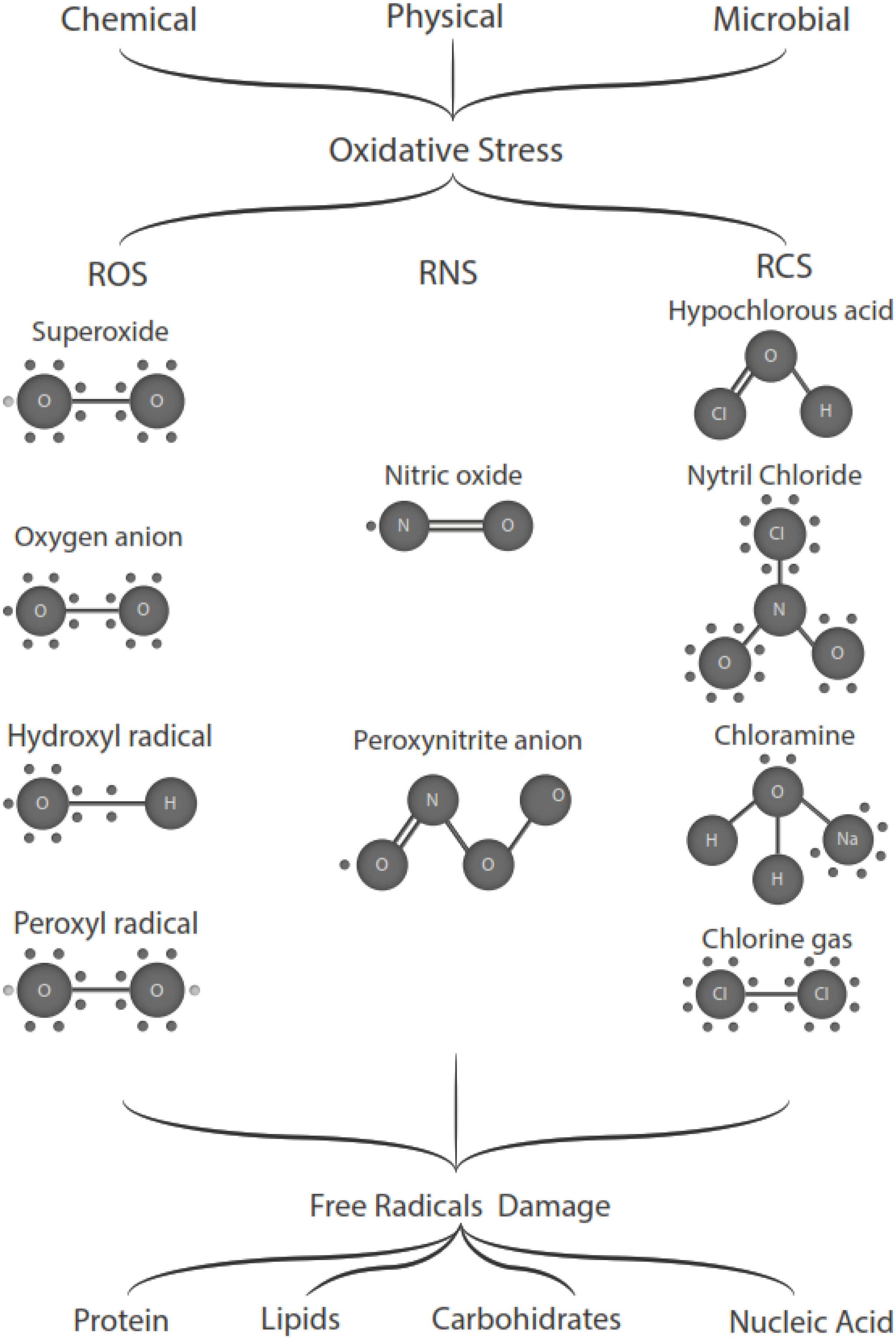
Free radicals are any chemical species that bear unpaired electron(s) in its outer orbit. Free radicals are highly reactive due to the presence of unpaired electrons. They readily participate in chemical chain reactions with lipids, proteins, complex carbohydrates, and nucleic acids.10 There are three different classes of reactive species relevant in biology, (1) Reactive oxygen species (ROS), derived from oxygen, represent the most critical class of radical species generated in living systems, including superoxide, oxygen anion, hydroxyl radical, peroxyl radicals; (2) Reactive nitrogen species (RNS), mainly nitric oxide (NO), peroxynitrite anion; (3) reactive chlorine species (RCS), including hypochlorous acid, nitryl (nitronium) chloride, chloramines, chlorine gas11,12 (Fig. 1). The internal biological sources of free radicals are mitochondria,13 xanthine oxidase,14 peroxisomes,15 and phagosome16; some external sources are cigarette smoking,17 environmental pollutants,18 and different types of radiation.19,20 Significantly, the intracellular concentration of ROS is transiently elevated in response to a stimulus, such as a cytokine, growth factors, or hormone. In physiological situations, where the antioxidant regulatory mechanisms rapidly control ROS release, but in infections or diseases, the increased oxidative stress is sustained or not counterbalanced, which overwhelms the antioxidant capabilities, leading to ROS induce tissue damage.8 Biological systems contain robust enzymatic and non-enzymatic antioxidant systems,21 such as reduced thiols, vitamins C and E, catecholamines, enzyme systems, as superoxide dismutase (SOD), peroxidase, catalase (CAT), and glutathione reductase, glutathione peroxidase, thioredoxin reductase, in an attempt to protect the integrity of cells and tissues, to counter the possible damage.9,22 For instance, it has been described that in experimental autoimmune encephalitis (EAE) exists a decline of the glutathione peroxidase levels, causing permanent damage.23

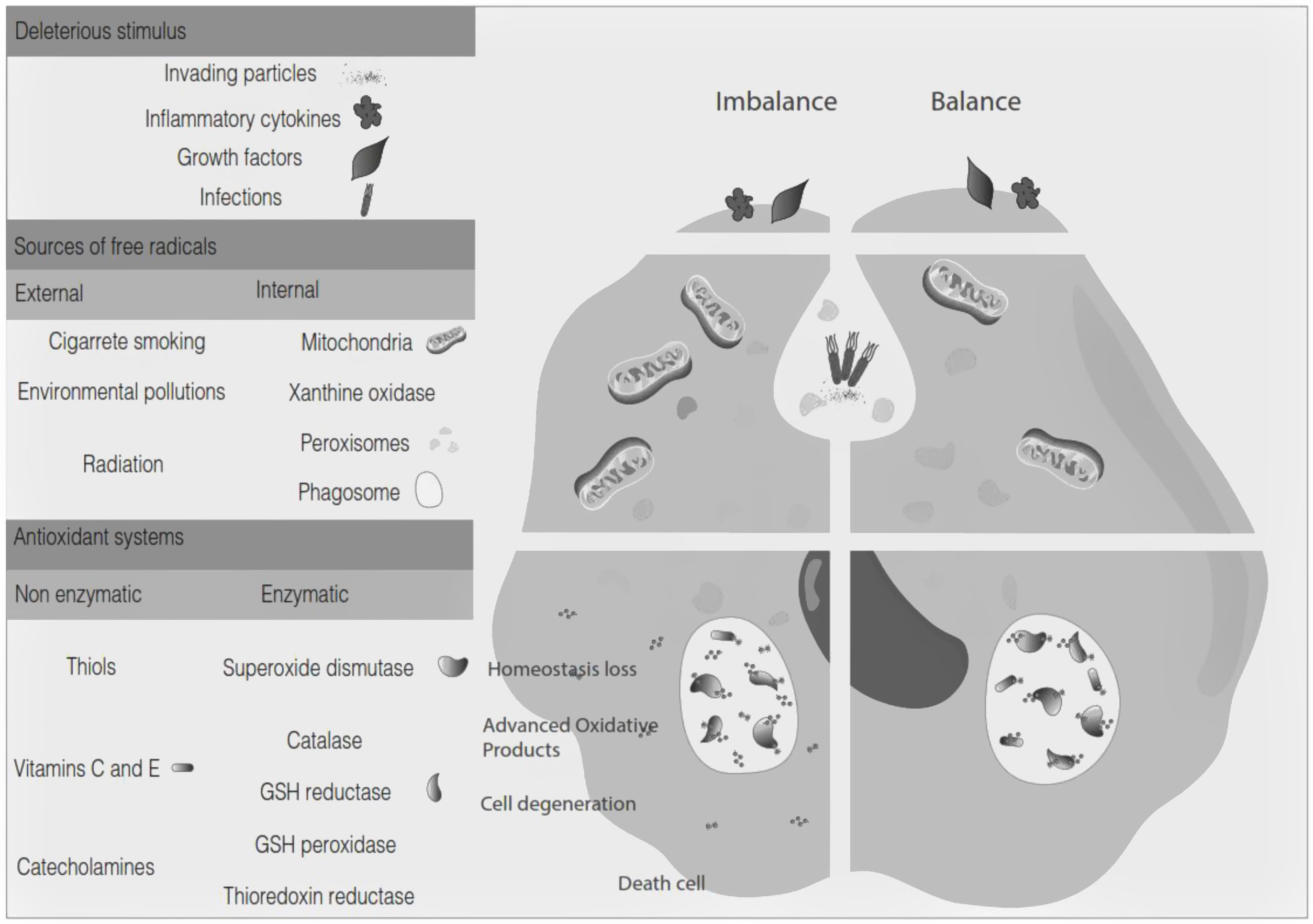
Oxidative stress denotes a shift in the prooxidant/antioxidant balance in favor of the first one. The delicate balance between antioxidants and the production of ROS could be compromised by exposure to toxic agents. For example, the release of inflammation precursors, hydroxyl anion, superoxide anion during phagocytosis of invading particles or microbes could exceed the antioxidant mechanisms and lead to oxidative and inflammatory damage to DNA, protein, or lipid oxidation9 (Fig. 2). The development of MS may be influenced by oxidative stress since it became an essential factor associated with demyelination. Furthermore, the brain tissue is susceptible to the action of radicals due to its high demand for oxygen and a limited possibility of obtaining antioxidants.24 In addition, cerebrospinal fluid (CSF) from MS patients showed elevated advanced product oxidation.25,26

Microglia is the specialized population of myeloid cells in the brain and spinal cord.27 It is considered a sentinel cell in the CNS, permanently sensing the environment, extending and retracting its projections to respond quickly to signs of invasion of pathogens or tissue damage.27 The microglia activated and mitochondrial dysfunction plays a role in oxidative processes. In the mature brain, microglia typically exist in a resting state characterized by ramified morphology and monitor the brain environment, such as brain injury or immunological stimuli; however, microglia are readily activated.28 Microglia activated by T cell release proteolytic enzymes, cytokines, oxidative products, and free radicals.24,29 The microglia activated uses pattern-recognition receptors (PRR) to identify neurotoxic stimuli, which stimulates NADPH oxidase activity, the principal mechanism through which microglia induce neurotoxicity.30,31
In MS, the microglia can acquire different phenotypes, depending on the nature and severity of the disease.27 Under demyelination and re-myelination conditions, microglia exhibit different phenotypes.27,32 This phenotypic heterogeneity of the microglia in MS is directly related to the temporal differences in the progression of lesions, together with the potential re-myelination or neurodegeneration and the interaction with other cell types.27,32
Macrophages and microglia in the M1 phenotype are pro-inflammatory, activated, and proliferates due to the interaction between the Toll-like receptors (TLR) receptor with ligands such as INF-γ or by stimulation of the same microglia with immunoglobulins or serum complement proteins.32,33 The microglia can change from an M1 phenotype to an M2 phenotype (anti-inflammatory) or to a mixed transitional state, where it co-expresses M1 and M2 markers, called M-Tran.32,33
Besides the production of extracellular ROS, NADPH oxidase generated superoxide and intracellular ROS that are common signaling mechanisms of phagocytes and regulate the expression of several pro-inflammatory functions of microglia.34 In general, the higher intracellular ROS, the more amplified the inflammatory response until either apoptosis or necrosis occurs.28
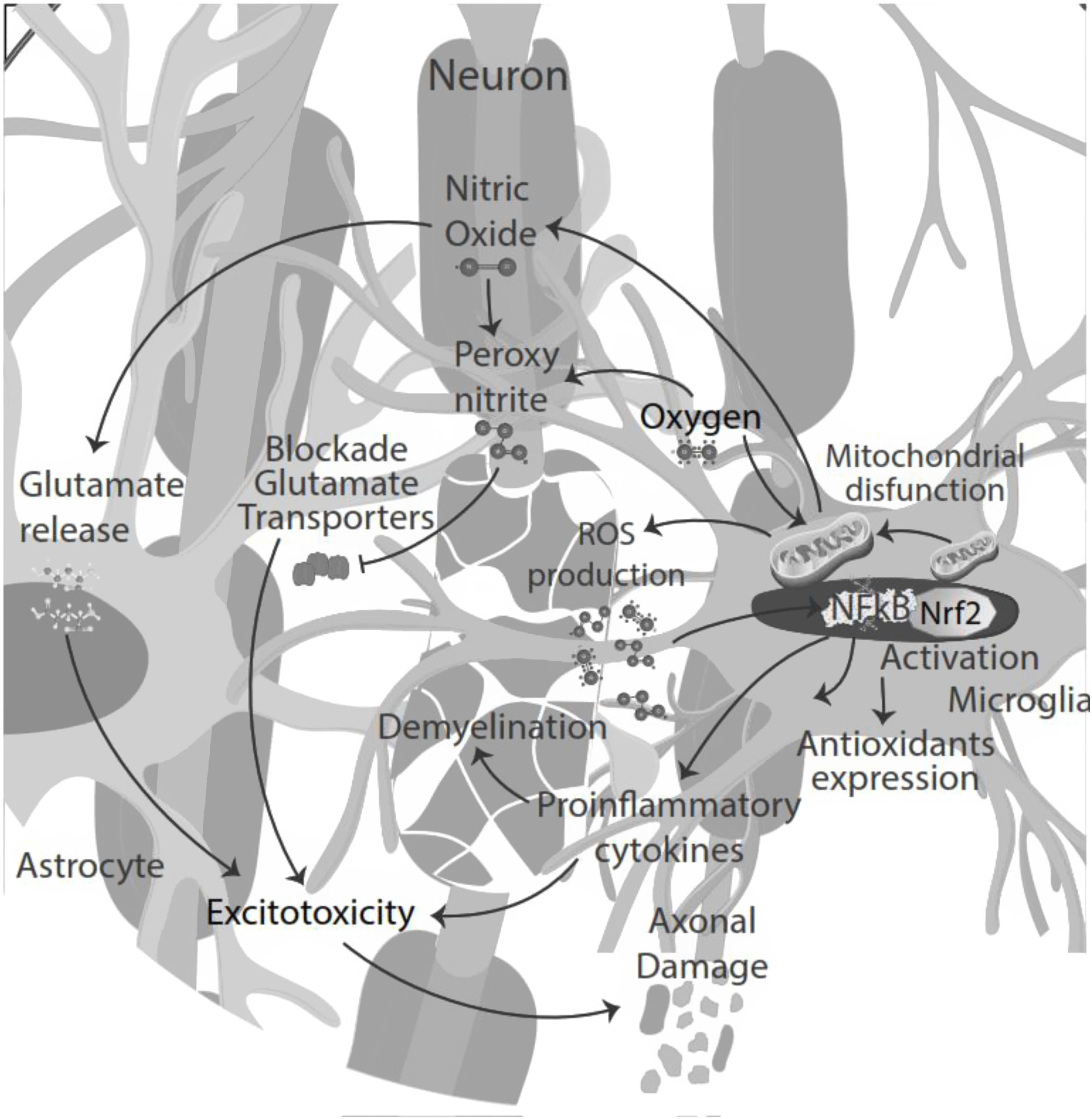
NO signaling inhibits microglia proliferation by activation of protein kinase-G.35 Also, NO secreted by microglia in an inflammatory microenvironment stimulates glutamate release from astrocytes, leading to excitotoxicity of neurons and glia. Excitotoxicity is the pathological process of damaged nerve cells or killed by abnormal stimulation by excitatory neurotransmitters. Decreased glutamate uptake by astrocyte transporters could also contribute to abnormal extracellular glutamate levels, directly toxic to oligodendrocytes, axons, and neurons. NO reacts with superoxide, producing peroxynitrite, which inactivates glutamate transporters in astrocytes resulting in neuronal necrosis or apoptosis by directly damaging myelin, oligodendrocytes, and axons.36
Astrocytes can significantly influence oxidative stress caused by the imbalance between ROS production and antioxidant defense mechanisms; the mitochondria are the primary source of cellular ROS.37 Oxidative stress and subsequent neuronal death due to ROS generation are correlated with cognitive dysfunction and alteration in memory and learning processes.37 Astrocytes are capable of releasing extracellular vesicles, thus reducing the levels of H2O2, ROS, and glutathione (GSH), increasing the activity of enzymes such as SOD and CAT.37 In patients with MS, up to five sub-clusters of neurons, six sub-clusters of oligodendrocytes, oligodendrocytes progenitors cells, astrocytes, pericytes, endothelial cells, and immune cells, have been identified as being involved in the pathogenesis of the disease.38
There is an important correlation between oligodendroglia and MS, including EAE, because these cells are responsible for the axonal myelination process in the CNS and are susceptible to death due to oxidative stress and ROS.38 At least six sub-clusters of oligodendrocytes are involved in MS; differences between sub-clusters of mature oligodendrocytes may indicate different MS lesions; this heterogeneity in altered oligodendrocytes may be substantial understanding the development and progression of the disease.27,38,39
Moreover, the presence of pro-inflammatory cytokines in the CSF and pro-oxidative markers led to cytokine-induced synaptic hyperexcitability and consequent glutamate-dependent neurotoxicity.24,40 Thus, oxidative stress and excitotoxicity seem to prevail and lead to an auto-toxic loop actively participating in neurodegeneration.41 During the inflammatory phase of the disease, T-cell infiltration could influence neuronal activity, contributing to glutamate excitotoxicity in neuronal cells42 (Fig. 3).

Mitochondria respond to oxidative stress by increasing their size and complexity, increasing energy production following acute damage. Active mitochondria produce high levels of ATP in high metabolic states and produce more ROS, and probably those high ROS levels damage the mitochondria and impede the production of ATP. Mitochondrial transportation is the earliest dysfunction in response to neuroinflammation and oxidative stress. A substantial decrease in the mean transport velocity was provoked by neuroinflammation and ROS. Thus, the impaired mitochondrial function could trigger several degenerative processes since the decrease ATP molecules available at these sites of strong demand like synapses, nodes of Ranvier, active growth cones, or axonal branches in neurons and glia.43
Oxidative stress induces transcriptional activation of various genes encoding antioxidant and detoxification enzymes through the nuclear factor-E2-related factor (Nrf2) antioxidant response element (ARE) pathway and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α). In the presence of oxidative stress, Nrf2 translocates to the nucleus, where activates ARE mediated gene transcription, such as SOD, glutathione peroxidase, catalase, heme oxygenases, quinone oxidoreductase 1 and 2, Parkinson disease-associated molecules Parkin and PINK and the mitochondrial antioxidants peroxiredoxin-3 and thioredoxin-2.44,45 The involvement of ARE, regulated enzymes in the development of the MS by the specific astrocyte Nrf2 activation prevents oligodendrocyte loss and demyelination, ameliorating intrinsic brain inflammation, and counteract axonal damage.46,47
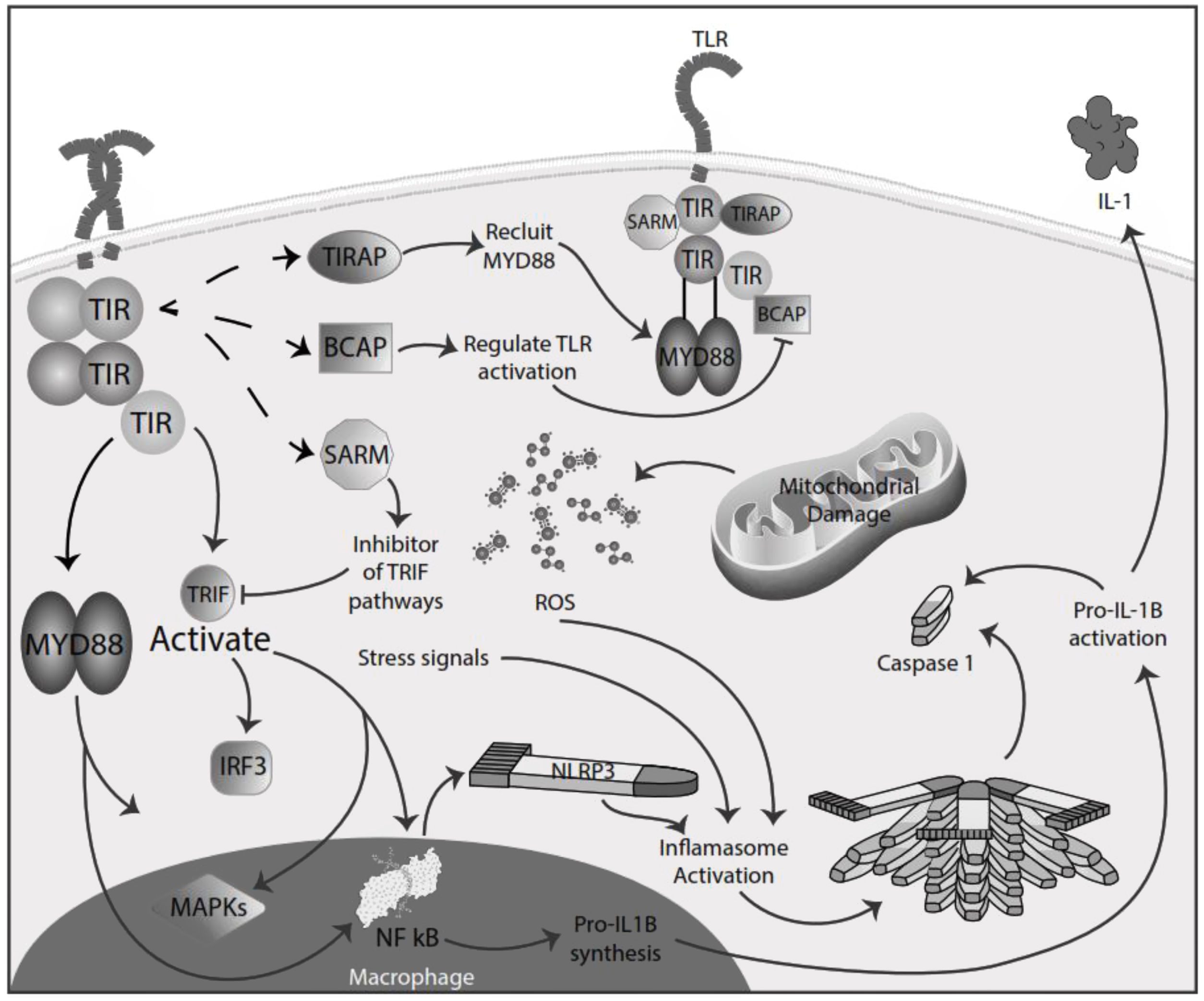
Inflammation in MSInflammation is commonly considered a complex reaction in the vascularized connective tissue in response to exogenous and endogenous stimuli. The ultimate goal of this protective response is to eliminate organisms, the initial cause of the cell injury, and the damage induced by the release of inflammatory mediators.48 However, there is much evidence that an exaggerated or unregulated prolonged inflammatory process can induce tissue damage promoting many chronic diseases, like neurodegenerative diseases. In an inflammatory condition, immune cells liberate ROS leading to oxidative stress.49,50 The ROS release can initiate an intracellular signaling cascade that enhances pro-inflammatory gene expression through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation in an early phase.51,52 Thus, inflammation and oxidative stress are closely related pathophysiological events tightly linked with one another.48 There are two major pathways by which the oxidative damage produced by free radicals promotes inflammatory responses: TLRs and the inflammasome53 (Fig. 4).

TLRs comprise PRR and are a family of type I transmembrane receptors characterized by an extracellular leucine-rich repeat domain; they can differentially recognize various pathogen-associated molecular patterns and an intracellular Toll/IL-1 receptor domain, which participates in TLR signal transduction.54,55 The TLR family consists of ten TLR1–TLR10,56 classified mainly into two subfamilies based on their localization, cell surface TLRs and intracellular TLRs. Cell surface TLRs include TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, whereas intracellular TLRs are localized in the endosome and include TLR3, TLR7, TLR8, and TLR9.57 TLRs also can recognize damage-associated molecular patterns (DAMPs). They are caused by numerous types of sterile stimuli, including mechanical trauma, ischemia, toxins, minerals, crystals, chemicals antigens, stress, and environmental pollution trigger pathogen-free inflammation state.58,59 TLR signaling is mainly divided into two pathways: the myeloid differentiation primary response 88 (MyD88) and toll/IL-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF) dependent pathways. Once activated any TLR by their ligands, it recruits members of a set of Toll/IL-1 receptor domain-containing adaptors such as MyD88, TRIF, toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP), TRIF-related adaptor molecule (TRAM), sterile-alpha and Armadillo motif containing protein (SARM), and B-cell adapter for PI3K (BCAP). MyD88 is utilized by all TLRs and activates NF-kB and mitogen-activated protein kinases (MAPK) signaling to induce inflammatory cytokine genes. TIRAP is a sorting adaptor that recruits MyD88 to the cell surface to TLR2, TLR4, and TLR9. TRIF is recruited to TLR3 and TLR4 and promotes an alternative pathway that activates interferon regulatory factor 3 (IRF3), NF-kB, and MAPKs to induce type I IFN and inflammatory cytokine genes. TRAM is selectively recruited to TLR4 but not TLR3 to link between TRIF and TLR4. SARM can act as a specific inhibitor of TLR3 and TLR4 signaling mediated by TRIF. BCAP is a regulator of TLR signaling60,61 (Fig. 4). In studies in the animal model for the study of MS, EAE has reported possible participation of these receptors in the physiopathology, also in MS patients has reported, especially TLR2 and TLR4, abnormal values in differents cell populations like B cell, also in sera.62,63 TLR2 may contribute to the development of MS through the reinforcement of Th1/Th17 cell-related responses, downregulation of regulatory T cells, inhibition of oligodendrocyte maturation, induction of poly ADP-ribose polymerase 1 – dependent pathway in microglia, macrophages, and astrocytes, and inhibition of type I interferons expression, besides induction of IL-17+γδ T cells such activated γδ T cells resemble IL-17+γδ T cells generated by the addition of IL-1β and IL-23. The presence of IL-17+γδ T cells results in toxic effects toward neurons that require a direct cell-cell contact between neurons and γδ T cells.64,65 Also, TLR2 and TLR3 activated the microglia, promoving the inflammation process and oxidative stress.66 In addition, the inflammatory role of TLR - MyD88 signaling has been described in MS, where it could be involved in the pathogenesis of MS and EAE by regulating the antigen presentation of dendritic cells and the integrity of the blood-brain barrier, and the activation of T cells and B cells. So, in addition to the maintenance of inflammation and oxidative stress production, these receptors could significantly participate in the disease's development and maintenance.67,68
The role of astrocytes in inflammatory processes can be both detrimental and beneficial69; astrocytes produce anti-inflammatory cytokines in response to inflammatory stimuli; conversely, the activation of astrocytes by pro-inflammatory cytokines induces IL-27.37,69
Inflammasomes are large cytosolic multiprotein complexes that assemble in response to detection of infection or stress-associated stimuli and lead to the activation of caspase-mediated inflammatory responses and initiation of an inflammatory form of cell death referred to as pyroptosis.70 There are two groups of specific inflammasomes based on the type of caspase involved: the classical, canonical inflammasome that triggers activation of caspase-1 directly, and the non-canonical inflammasome, which uses other caspases, like caspase-4, caspase-5, and caspase-8, to convey inflammation.71 The inflammasomes are conformed by a sensor receptor, like PRR; an adaptor protein; and an effector enzyme, like caspase. All components catalyze a cellular reaction to protect against an immediate danger via cytokine secretion and cell death.70 Families of PRR can be divided into two groups, the transmembrane PRR, like TLRs and C-type lectin families; and the cytoplasmic PRR, like the nucleotide-binding and oligomerization domain (NOD)-like receptors (NLR), retinoic acid-inducible gene-I (RIG1)-like receptors (RLR) and absent-inmelanoma (AIM)-like receptors (ALR).72 Formation of the NLR family pyrin domain containing 1 (NLRP1), NLRC4, and AIM2 inflammasomes is promoted by specific stimuli, while the NLRP3 inflammasome is the most studied and is activated in response to several physically and chemically diverse triggers, like stress signals.73 The adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) interacts with the upstream inflammasome sensor molecules via the pyrin domain.74 This interaction triggers the assembly of ASC into oligomers through homotypic PYD–PYD interactions into a large filamentous scaffold. Inactive procaspase-1 monomers are recruited to ASC filaments through CARD-CARD interactions. Therefore, they are brought into proximity for optimal self-activation. ASC is a bipartite molecule containing both a PYD and a CARD, enabling it to bridge the sensors (NLRs or ALRs) and the effector pro-caspase-1.74,75 In a canonical pathway, pro-caspase 1 is activated, leading to the cleavage of pro-IL-1β and pro-IL-18 and the generation of the mature biologically active cytokines.74–76 ROS has been widely implicated in NLRP3 activation, and many NLRP3 activating stimuli, including ATP, alum, uric acid, and nigericin, all induce more ROS production.77 Although ROS's role in NLRP3 inflammasome activation remains controversial because intracellular ROS produced by the NADPH oxidase system was thought to activate NLRP3, NADPH oxidases inhibitors inhibit inflammasome activation.70,78,79
The inflammasome contributes to the inflammatory response in MS. For example, inflammasome-associated caspases mediate the maturation and release of the pro-inflammatory cytokines IL-1β and IL-18 and activate the pore-forming protein Gasdermin D; this process contributes to neuroinflammation and demyelination.80 Also, NLRP3 inflammasome is activated and contributes to cognitive deficits in EAE mice, and it may act via regulating astrocyte phenotype alteration.81 Some genetic studies have described that constitutive activation of NLRP3 inflammasome could represent a risk factor for MS clinical presentation.82,83 Due to these results, inflammasome components have been studied as biomarkers of the development of the disease.84
Both TLRs and the inflammasome promote inflammatory responses, leading to the release of pro-inflammatory cytokines. However, inappropriate pro-inflammatory cytokines release can drive sterile inflammation and increase oxidative stress, creating a vicious circle; the release promotes other production. All of these mechanisms contribute to the development of the inflammatory process in MS.
ConclusionsOxidative stress is the imbalance between reactive species and antioxidant systems. Increased oxidative stress can induce damage to the cellular structure and potentially destroy tissues. The relationship between inflammation and oxidative state is close because the stress could be initiated by the signaling of TLRs and the inflammasome. Inflammation could contribute to the ROS and RNS production, and vice versa, establishing an auto-toxic loop that helps develop a pathophysiologic mechanism in diseases like MS.
Microglia activated is the principal ROS producer, increasing the inflammatory environment until apoptosis, necrosis, or pyroptosis occurs. High amounts of ROS could act over mitochondria impeding the ATP production necessary for neurons and glia's regular activity. Furthermore, RSN can induce excitotoxicity through the glutamate release and impair its uptake system. The inflammasome contributes to the establishment of an inflammatory state, releasing pro-inflammatory cytokines and increasing oxidative stress. All of these mechanisms participate in the development and progress of MS.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestNone.

