



La sobrepoblación y el crecimiento industrial causan un aumento en la contaminación del aire, principalmente de partículas suspendidas y en la formación de ozono. La exposición repetida a bajas dosis de ozono, como la de un día con alta contaminación del aire, genera un estado de estrés oxidativo crónico, el cual causa pérdida de espinas dendríticas, alteraciones en la plasticidad cerebral y en los mecanismos de aprendizaje y memoria, así como muerte neuronal y pérdida de la capacidad de reparación cerebral. Esto tiene un impacto directo en la salud humana, aumentando la incidencia de enfermedades crónico-degenerativas.
DesarrolloSe realizó una búsqueda de artículos originales y revisiones en PubMed, Scopus y Google Scholar (2000-2018) sobre las principales consecuencias de la exposición a ozono, sobre la plasticidad sináptica, sobre el procesamiento de la información en los procesos cognitivos y sobre las alteraciones que pueden llevar al desarrollo de enfermedades neurodegenerativas.
ConclusionesEsta revisión describe uno de los mecanismos fisiopatológicos del efecto de la exposición repetida a bajas dosis de ozono, a través de producir un estado de estrés oxidativo crónico, lo cual causa pérdida de la plasticidad sináptica; esta función cerebral es clave tanto en el procesamiento de información como en la generación de cambios estructurales en las poblaciones neuronales. También se aborda el efecto de la exposición crónica a ozono sobre el tejido cerebral, y la estrecha relación entre la contaminación por ozono y la aparición y evolución de las enfermedades neurodegenerativas.
Overpopulation and industrial growth result in an increase in air pollution, mainly due to suspended particulate matter and the formation of ozone. Repeated exposure to low doses of ozone, such as on a day with high air pollution levels, results in a state of chronic oxidative stress, causing the loss of dendritic spines, alterations in cerebral plasticity and in learning and memory mechanisms, and neuronal death and a loss of brain repair capacity. This has a direct impact on human health, increasing the incidence of chronic and degenerative diseases.
DevelopmentWe performed a search of the PubMed, Scopus, and Google Scholar databases for original articles and reviews published between 2000 and 2018 and addressing the main consequences of ozone exposure on synaptic plasticity, information processing in cognitive processes, and the alterations that may lead to the development of neurodegenerative diseases.
ConclusionsThis review describes one of the pathophysiological mechanisms of the effect of repeated exposure to low doses of ozone, which causes loss of synaptic plasticity by producing a state of chronic oxidative stress. This brain function is key to both information processing and the generation of structural changes in neuronal populations. We also address the effect of chronic ozone exposure on brain tissue and the close relationship between ozone pollution and the appearance and progression of neurodegenerative diseases.
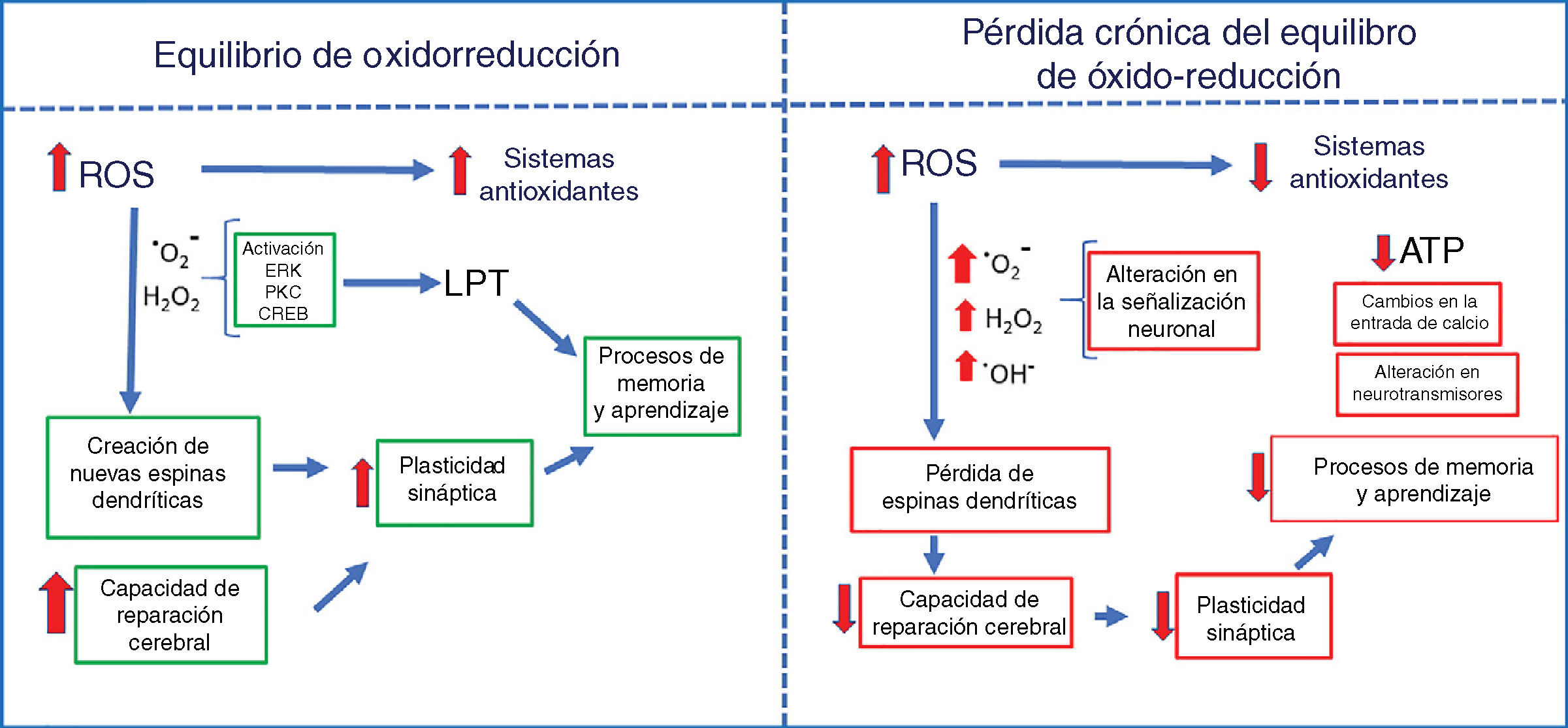
La exposición repetida a bajas dosis de ozono tal como sucede durante los días con alta contaminación genera en el organismo un estado de estrés oxidativo crónico. El efecto de la radiación solar sobre los gases contaminantes (el monóxido y el dióxido de carbono, el óxido de nitrógeno y los compuestos orgánicos volátiles) produce moléculas altamente oxidantes, como el ozono. La exposición crónica de este gas genera un estado de estrés oxidativo, el cual produce efectos adversos a bajas concentraciones, tales como un aumento en la producción de especies reactivas de oxígeno (ROS por sus siglas en inglés, reactive oxygen species) en el organismo y un déficit en la respuesta de los sistemas antioxidantes. El estado de estrés oxidativo lo podemos definir como un desequilibrio entre la tasa de producción de moléculas oxidantes y la degradación de estas por los sistemas de defensa antioxidantes1. En un equilibrio redox, las señales oxidantes participan como señalizadores tanto extra como intracelulares que regulan el metabolismo celular, así como la respuesta inmune. Los animales aerobios, principalmente los vertebrados, obtienen su energía de la oxidación de sustratos orgánicos (glucosa) usando el oxígeno molecular. Sin embargo, algunas veces el oxígeno se reduce parcialmente, dando origen a moléculas prooxidantes como el ion superóxido y algunos compuestos intermedios muy reactivos2. Estos intermediarios pueden ser fácilmente neutralizados por los sistemas antioxidantes manteniendo el balance de óxido-reducción. No obstante, un aumento en las especies reactivas y una falla en el sistema de antioxidantes producen un estado de estrés oxidativo crónico3 que causa alteraciones en procesos celulares como el daño de organelos, alteraciones metabólicas y el inicio de la apoptosis1 (fig. 1).
 y durante una pérdida crónica del equilibrio de oxidorreducción (derecha).")
Las ROS incluyen los radicales libres de oxígeno (RL) y sus metabolitos originados en la célula, tales como los radicales hidroxilo y superóxido, el peróxido de hidrógeno (H2O2), etc. Estas ROS son capaces de oxidar el ADN4,5, las proteínas y los lípidos5,6.
Las ROS se pueden producir a través de 2 vías: la endógena y la exógena. En la vía endógena, las ROS son producidas durante el metabolismo aerobio mitocondrial, en el retículo endoplásmico y en los peroxisomas7. Además, el aumento en la producción de ROS va acompañado de especies reactivas de nitrógeno, mientras que en la vía exógena se generan como resultado de la exposición a varios factores ambientales, como: a) la contaminación del aire y los oxidantes obtenidos de la combustión de los hidrocarburos y del tabaco ?estos últimos interactúan con moléculas de la piel e incluso la penetran hasta llegar a la circulación sanguínea8,9?, y b) la radiación UV proveniente del sol, que es absorbida por la piel y es capaz de inducir la formación de una variedad de prooxidantes10.
Especies reactivas de oxígenoEl oxígeno ha sido considerado un elemento esencial para el metabolismo de los organismos aerobios. Sin embargo, bajo ciertas circunstancias, este gas puede llegar a ser tóxico. En el metabolismo, el oxígeno se integra a las reacciones químicas para la obtención de energía en la mitocondria y se producen los RL, los cuales son neutralizados por los sistemas antioxidantes como la superóxido dismutasa (SOD) y el sistema glutatión, a través de una serie de reducciones de diferentes reacciones químicas, mediante la transferencia de electrones hasta producir agua11,12.
El impacto que producen las ROS está en función de su concentración, es decir, en concentraciones fisiológicas durante un equilibrio redox, son señalizadoras de diferentes respuestas metabólicas celulares; el aumento de las ROS estimula las defensas antioxidantes y el crecimiento celular, regula el ciclo celular, modula el sistema inmune, etc.13,14. Sin embargo, a bajas concentraciones como consecuencia de la exposición crónica al ozono, las defensas celulares antioxidantes producen un déficit en la respuesta de los sistemas antioxidantes, lo que causa una alteración en la señalización intracelular, así como una alteración en los procesos metabólicos y muerte celular7,15–17. Cabe destacar que dentro de la señalización intracelular también las señales oxidantes tanto a niveles fisiológicos como a altas concentraciones inducen la respuesta antioxidante endógena; sin embargo, la exposición repetida a bajas dosis produce una incapacidad de los sistemas antioxidantes para contender con el estrés oxidativo crónico. Esto explicaría el efecto dual del ozono: por un lado, el uso terapéutico del ozono administrado en forma aguda a altas dosis, y por el otro, su relación con enfermedades degenerativas cuando se administra crónicamente a bajas dosis.
Defensas antioxidantesDurante la evolución de la vida, la aparición de los pigmentos dio origen a los organismos fotosintéticos, los cuales produjeron un aumento en los niveles de oxígeno en forma de gas en la atmósfera terrestre, permitiendo el desarrollo de los mecanismos necesarios para utilizar esta molécula oxidando la glucosa y obtener gran cantidad de energía en forma de ATP y generando ROS; esto permitió a las células tener sistemas de producción de energía altamente eficientes. Esta ventaja evolutiva de los organismos aerobios también causó el aumento en la producción de RL y de las ROS, lo que llevó a la evolución de sistemas de defensa antioxidantes12.
Estos sistemas de defensa están constituidos por sustancias de bajo peso molecular capaces de neutralizar espontáneamente a las ROS y a sus productos derivados, y pueden actuar de las siguientes formas: a) disminuyendo la concentración de moléculas oxidantes; b) evitando la iniciación de la reacción al «barrer» (cubrir o detener una reactividad química muy alta) los primeros RL que se forman; c) uniéndose a los iones metálicos para evitar la formación de ROS; d) transformando los peróxidos en productos menos reactivos, y e) deteniendo la propagación y el aumento de RL12,18. Los sistemas antioxidantes ayudan a mantener el equilibrio redox como una barrera molecular que protege contra el daño oxidante19.
Por otro lado, las proteínas que se unen a los metales de transición, tales como la transferrina, la ferritina y la ceruloplasmina, proporcionan protección debido a que limitan la capacidad del metal para participar en las reacciones de oxidación20; a esto se añade la existencia de enzimas endógenas específicas que previenen la formación de especies oxidantes, como las SOD, la catalasa, la glutatión reductasa y la glutatión peroxidasa, entre otras. Dichas enzimas actúan como las principales defensas antioxidantes contra los oxidantes primarios; además, numerosas enzimas antioxidantes, incluyendo enzimas lipolíticas, enzimas proteolíticas y glutatión S-transferasas, pueden actuar por barrido y por la eliminación de las ROS. Así, en condiciones fisiológicas, las células son capaces de hacer frente a la afluencia de concentraciones excesivas de ROS equilibrando su producción e inactivación21–23.
También los antioxidantes ingeridos en la dieta, como el β-caroteno (precursor de la vitamina A), el ácido retinoico y el ácido ascórbico (vitamina C), neutralizan y «barren» el O2−•, y mantienen al α-tocoferol (vitamina E) en su estado reducido y activo. El α-tocoferol detiene la cadena de reacciones de la peroxidación lipídica y el glutatión, y otros tioles (por ejemplo, el dihidrolipoato) bloquean los H2O2 y contribuyen al mantenimiento del tocoferol y del ácido ascórbico en sus formas reducidas12,24–26.
Contaminación y modelos de estudio de estrés oxidativo causado por las especies reactivas de oxígenoActualmente existen 2 tipos de aproximaciones experimentales en el estudio del estrés oxidativo: la producción de especies reactivas de forma exógena y la endógena. En la primera, si se usa un modelo animal, este es expuesto a ambientes oxidantes como puede ser la exposición repetida a dosis semejantes a contaminación ambiental, como, por ejemplo, la que se usa en animales expuestos de forma repetida a bajas dosis de ozono7,16,27 o la administración de sustancias tóxicas, como el paraquat, que inducen el estado de estrés oxidativo crónico mediante la generación de RL28,29 y la pérdida de las defensas antioxidantes. En la segunda, la producción del estrés oxidativo requiere la sobreproducción de moléculas prooxidantes, como ocurre en el modelo del ácido 3-nitropropiónico, el cual inhibe la cadena respiratoria mitocondrial y causa un déficit de ATP, llevando a un estado de estrés oxidativo o a una inhibición en la producción de antioxidantes30,31, modelos transgénicos32–34.
Además, se han utilizado varias especies animales como modelos de estudio del efecto de las ROS; tal es el caso de invertebrados como la mosca de la fruta Drosophila melanogaster y el nematodo Caenorhabditis elegans, en los que se han inducido mutaciones puntuales en genes específicos para modificar la cantidad de moléculas de la SOD y la catalasa disponibles en los diferentes tejidos35–37. También se han desarrollado otros modelos de inducción del estrés oxidativo en mamíferos, como ratas, ratones y monos. A partir de esos diferentes modelos, se han desarrollado aproximaciones tanto in vitro como in vivo.
Con los datos obtenidos de los modelos experimentales in vitro e in vivo se ha podido dilucidar cómo el estrés oxidativo puede producir una gran variedad de efectos tóxicos; entre estos, son particularmente relevantes los efectos neurotóxicos, los cuales muestran disfunción neuronal, degeneración y muerte celular.
Por otra parte, los cultivos celulares permiten medir directamente los cambios del estado oxidante de las neuronas, lo cual hace posible estudiar la participación del estrés oxidativo en el desarrollo de enfermedades38–40. Otra aproximación metodológica es el estudio del aumento en la producción de los RL por medio de una prueba molecular fluorescente sensible a oxidación, como la diclorodihidrofluoresceína, permitiendo así la detección de la producción de las ROS intracelulares41–43. Esta técnica ha sido utilizada en el cultivo primario de neuronas del hipocampo de rata, en donde se han identificado moléculas prooxidantes mediante microscopia de fluorescencia44.
Es importante señalar que el desarrollo de un modelo animal, en el cual se puedan replicar las condiciones de contaminación ambiental a las que el ser humano puede estar expuesto, ha permitido establecer que un factor crucial es el establecimiento de un estado crónico de estrés oxidativo, el cual lleva a una alteración en la señalización celular, causando pérdida de la regulación de la respuesta inflamatoria y alteración de vías de señalización intracelular asociadas a la regulación de las funciones celulares. Todo lo anterior forma parte de los mecanismos complejos involucrados en los procesos neurodegenerativos. Además, la exposición crónica a bajas dosis de ozono es un modelo no invasivo que permite el estudio de la participación del estrés oxidativo en los procesos neurodegenerativos y que, por sí mismo, sin otro factor agregado, es capaz de causar un proceso de neurodegeneración progresiva en el hipocampo de ratas expuestas a dicho gas45. El estrés oxidativo causado por la exposición a oxígeno produce un aumento en las ROS7, que inducen daño celular46 y la peroxidación lipídica en diferentes regiones cerebrales45,47. También se presenta disfunción mitocondrial48, daño al retículo endoplásmico16, aumento de la microglía activada45, incremento de la interleucina IL-17A17 y muerte neuronal dependiendo del tiempo de exposición45,49.
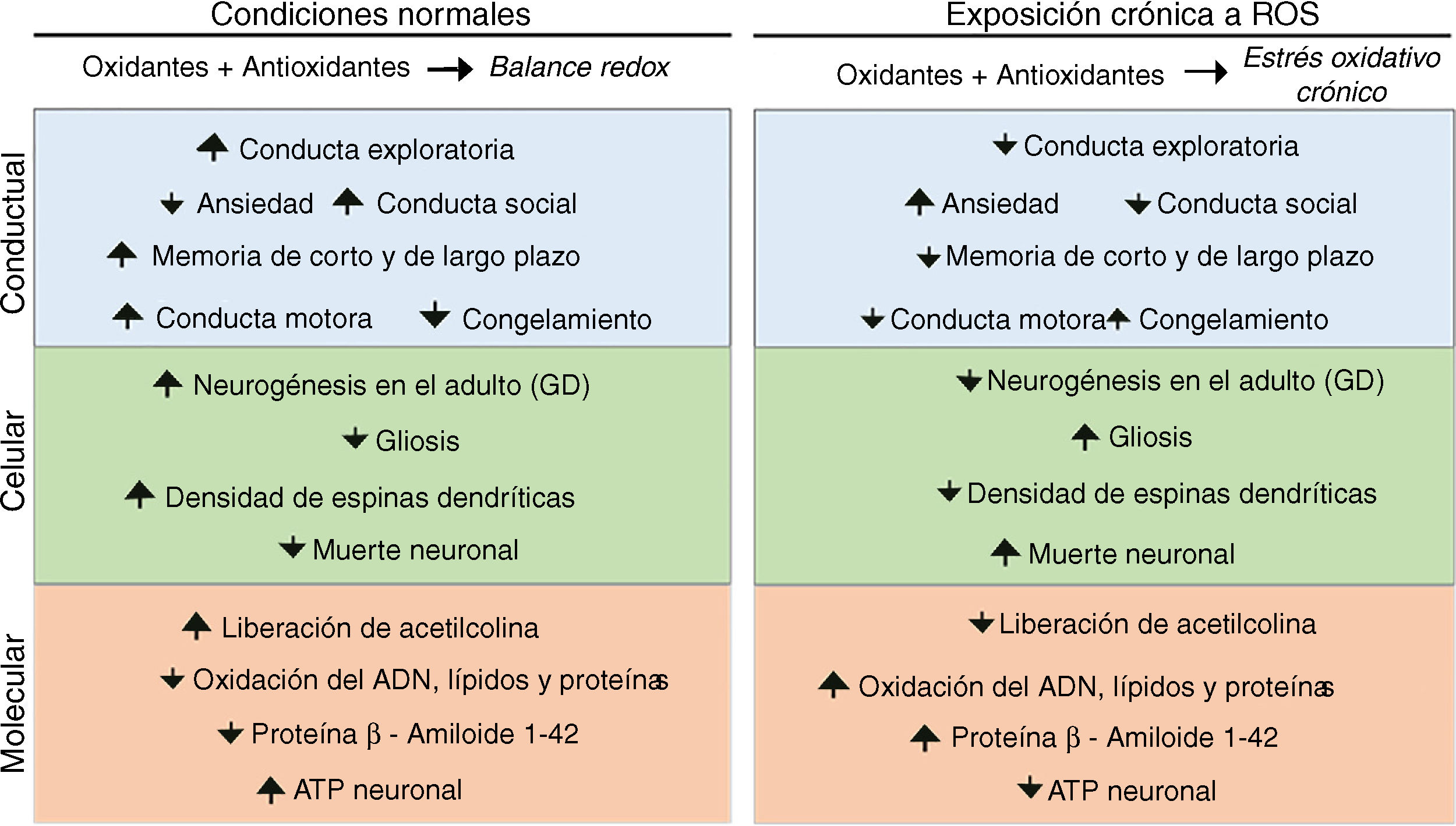
Estrés oxidativo, plasticidad sináptica y enfermedades neurodegenerativasEl estrés oxidativo crónico producido principalmente por la exposición crónica a ozono afecta el desempeño conductual y la cognición. Se ha observado que la exposición crónica a ozono induce en roedores una disminución en la actividad exploratoria y un aumento en la conducta de congelamiento50,51; asimismo, se ha reportado una disminución en el porcentaje del tiempo de permanencia en los brazos abiertos en el laberinto elevado, una disminución del tiempo de interacción social y una disminución en la conducta de escalada52. También se ha visto un deterioro en las memorias de corto y de largo plazo en la tarea de evitación inhibitoria15 (fig. 2).

A nivel celular, la exposición crónica a ozono inhibe la neurogénesis en el giro dentado del animal adulto; también promueve la gliosis y un aumento de las microglías fagocíticas45. Colín-Barenque et al. estudiaron el efecto de la exposición crónica a ozono y encontraron una disminución significativa en la densidad de espinas dendríticas en el bulbo olfatorio de ratas desde la primera exposición, conservándose el efecto hasta los 10 días53. También se ha reportado una disminución en la densidad de espinas de las dendritas secundarias y terciarias de las neuronas medianas espinosas del estriado y las neuronas piramidales de la corteza prefrontal50. A nivel de los sistemas de neurotransmisión, la exposición crónica a ozono produce una disminución en los niveles de acetilcolina, acetilcolinesterasa y acetilcolintransferasa en el área CA3 del hipocampo dorsal15 (fig. 2).
Algunas ROS juegan un papel esencial en la señalización relacionada con la plasticidad sináptica en varias regiones cerebrales. En el hipocampo, el anión superóxido es requerido para la activación de proteínas cinasas como la cinasa regulada por señal extracelular y la proteína cinasa C durante la inducción de la LTP54. En cambio, la presencia de los antioxidantes y de los barredores del anión superóxido impide la activación de la cinasa regulada por señal extracelular en rebanadas de hipocampo55; esto implica el bloqueo de varias cascadas de señalización que desencadenan la expresión génica asociada a la plasticidad sináptica.
Otros estudios muestran que en el hipocampo, el H2O2 es fundamental para la fosforilación de la cinasa regulada por señal extracelular y del elemento de unión reactivo a cAMP en respuesta a la estimulación del receptor NMDA56,57. También participa en la liberación de Ca+2 de los reservorios intracelulares, lo que va a permitir que se activen cascadas de señalización que promueven la expresión de genes que participan en el procesamiento de información. Esto podría explicar por qué el uso de ozono y de otros agentes prooxidantes a altas dosis como terapia regenerativa en un intervalo temporal corto es benéfico para la salud neuronal, siempre y cuando los sistemas antioxidantes estén activos. No obstante, también se ha observado que el ozono a bajas dosis causa un exceso en la producción de los agentes oxidantes debido al déficit de las defensas antioxidantes, lo cual es altamente tóxico para los procesos de plasticidad sináptica, que normalmente van a incidir directamente en problemas cognitivos.
El destino de una sinapsis cerebral depende del balance redox. Si el ambiente celular es prooxidante, las ROS producirán una poda de espinas dendríticas; en cambio, si el ambiente celular es antioxidante, el efecto de las ROS será neutralizado y la sinapsis se conservará58.
Si bien hay evidencias sobre la incidencia de las ROS sobre algunas moléculas que participan activamente en procesos plásticos, existe muy poca información acerca del efecto de estas moléculas prooxidantes sobre los cambios en la estructura neuronal que subyace a la plasticidad sináptica. Ciertas áreas del cerebro humano, como los ganglios basales y la sustancia negra (SN), contienen grandes cantidades de hierro total y otros metales de transición que constituyen factores cruciales en la generación de ROS59,60.
El papel de las especies reactivas de oxígeno en la enfermedad de AlzheimerLos estudios realizados en cultivos primarios de células corticales de rata mostraron que la proteína β-amiloide induce deficiencias tanto en el complejo i (NADH deshidrogenasas) como en el complejo iv (citocromo c oxidasa). El complejo i es considerado el principal promotor de ROS en el estado fisiológico normal y se ha indicado que alteraciones fundamentales en su funcionamiento pueden resultar en una mayor generación de ROS e inducir el consumo total de la energía, como resultado de la interrupción de la fosforilación oxidativa. Lesiones mitocondriales causadas por la sobreproducción de la proteína β-amiloide en la enfermedad de Alzheimer (EA) inducen la generación de ROS, lo cual resulta en un daño y, posteriormente, en la muerte celular61,62. El agotamiento del ATP neuronal produce un mal funcionamiento de la neurotransmisión y altera el transporte axonal. Esto afecta a los canales iónicos dependientes de ATP, alterando así el equilibrio iónico citosólico61.
En roedores, se ha demostrado que el estado de estrés oxidativo causa en el hipocampo un proceso de neurodegeneración progresiva que induce muerte neuronal, pérdida de la neurogénesis, estimulación glial y aumento de la síntesis del β-amiloide 1-42, además de cambios conformacionales en dicha molécula27,45.
Por otra parte, en algunos modelos animales y humanos se ha demostrado un aumento prolongado del estado prooxidante durante todo el curso de la EA, lo cual se apoya en varias evidencias que involucran a las ROS como causa de los cambios en el estado redox del cerebro envejecido y que podrían contribuir a la patogénesis de la EA63–65. El debilitamiento de la membrana fosfolipídica, como principal resultado de la lipoperoxidación, ha sido reconocido como una de las causas en las enfermedades neurodegenerativas, como la EA66. La proteína β-amiloide acentúa la capacidad de iniciación de la peroxidación lipídica. El aumento en la incidencia de los peróxidos lipídicos y la disminución de la actividad de las enzimas antioxidantes se encuentran fuertemente correlacionados con la formación de placas seniles y ovillos neurofibrilares en el cerebro con EA. Además, marcadores de estrés oxidativo han sido detectados en el tejido cerebral y en el líquido cefalorraquídeo en pacientes con EA67,68.
En la enfermedad de Parkinson (EP), se ha demostrado que el estado de estrés oxidativo está ampliamente asociado a un aumento en la oxidación de la dopamina y el hierro, y a un déficit del sistema glutatión, lo cual causa un círculo vicioso que mantiene el estado de estrés oxidativo y la destrucción de la neurona dopaminérgica46,69. En la EP está implicada la acumulación de α-sinucleína, así como la pérdida gradual de las neuronas dopaminérgicas en la SN70,71. En esta enfermedad se ha observado un aumento en los niveles de 8-hidroxideoxiguanosina, asociado con un incremento en las deleciones que se presentan comúnmente en el ADN mitocondrial de las neuronas dopaminérgicas no afectadas en la SN y en el suero72,73. Además, se ha encontrado una reducción en la concentración de ácidos grasos libres poliinsaturados, mientras que aumentan los marcadores de peroxidación de lípidos (malondialdehído y 4-hidroxinonenal, entre otros)74,75. También se ha confirmado la carbonilación de proteínas como resultado de daño oxidante76; estos cambios no son patognomónicos de la EP, ya que también están presentes en otras enfermedades neurodegenerativas como consecuencia de la presencia del estado de estrés oxidativo.
Estudios del daño del ADN mitocondrial han contribuido al desarrollo de nuevas hipótesis sobre la etiología de algunas enfermedades degenerativas relacionadas con la edad. Varias investigaciones refieren un déficit en la actividad enzimática del complejo i mitocondrial en la SN y la corteza prefrontal de pacientes con EP, por lo que parece ser un defecto específico del área77,78. Este cambio específico en el complejo i en el estriado y la SN también se ha descrito en modelos animales y en los pacientes con EP79,80.
La esclerosis lateral amiotrófica es una alteración letal y degenerativa de las motoneuronas y se ha demostrado que está relacionada con una mutación en el cromosoma 21 que codifica para la SOD de Cu/Zn, pero esta alteración solo se presenta en la forma familiar de la enfermedad y se transmite de forma autosómica dominante81. Este aspecto brinda la posibilidad de un mayor acercamiento, desde el punto de vista terapéutico, entre este grupo de pacientes y la gran mayoría de los enfermos con esclerosis lateral amiotrófica que no presentan carácter hereditario. Con el propósito de obtener más información sobre el posible papel de la actividad de la SOD de Cu/Zn en esta enfermedad, se produjeron ratones transgénicos que sobreexpresan el gen de la SOD Cu/Zn humano82. La alteración en los animales condujo a cambios patológicos en las motoneuronas, como la pérdida y destrucción de axones terminales y el desarrollo de múltiples terminales muy pequeñas83. La toxicidad que genera un exceso en la actividad de la SOD puede explicarse por la formación de H2O2, el cual es el precursor del radical OH•. El radical OH• puede reaccionar con gran variedad de moléculas, en particular con los ácidos grasos poliinsaturados, y, finalmente, genera la reacción en cadena de la peroxidación lipídica y la destrucción de la integridad de las membranas84.
Existen otras evidencias de cómo el estrés oxidativo desempeña una importante función en la patogénesis de la esclerosis lateral amiotrófica, tanto esporádica como familiar. Se ha observado que las ROS generadas en el curso de la muerte celular y neuronal pueden ser moduladas por la actividad antioxidante del protooncogén Bcl-285. Se ha propuesto la excitotoxicidad del glutamato como mecanismo fisiopatológico en esta enfermedad86,87. En el curso de este proceso, pueden generarse los O2−• a través de varias vías, que incluyen la producción directa de ROS, la activación de la xantina oxidasa mediada por el calcio y la iniciación de la cascada del ácido araquidónico por la fosfolipasa A288–90. Finalmente, el NO•, que también es producido por esta vía, puede interactuar con el anión O2−• para formar los ONOO−, que son especies altamente reactivas88.
ConclusionesLa contaminación del aire por ozono induce un estado crónico de estrés oxidativo. El estado crónico de estrés oxidativo es un factor crucial en las enfermedades crónico-degenerativas, que afectan la calidad de vida. El desarrollo de un modelo no invasivo animal para el estudio del estrés oxidativo crónico que replica las condiciones medioambientales en las que nos encontramos actualmente, que implica la exposición crónica a bajas dosis de ozono y la inhibición de la capacidad de respuesta de los sistemas antioxidantes, puede ayudar a dilucidar la asociación entre el estrés oxidativo y el desarrollo de las enfermedades crónicas degenerativas, provee información importante acerca de las interacciones moleculares y celulares que afectan diferentes estructuras cerebrales e inducen enfermedades neurodegenerativas.
El incremento crónico de las ROS, acompañado de un déficit de los sistemas antioxidantes, representa un riesgo importante en el desarrollo de enfermedades neurodegenerativas, ya que el efecto de estas moléculas prooxidantes incide directamente sobre la plasticidad sináptica, específicamente en la pérdida de espinas dendríticas, la muerte neuronal y la pérdida de la capacidad de reparación cerebral, lo cual conlleva la pérdida de contactos sinápticos, afectando la capacidad del procesamiento de información por parte de las redes neuronales; por otro lado, también ocurren alteraciones en cascadas de señalización que inciden en la expresión génica, causando un déficit cognitivo que puede desencadenar la aparición de enfermedades neurodegenerativas.
FinanciaciónEl presente trabajo fue financiado por el Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT-IN221417) y el Consejo Nacional de Ciencia y Tecnología (CONACyT 219703).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos a la Dirección General de Asuntos del Personal Académico (DGAPA) de la Universidad Nacional Autónoma de México por la asignación de la beca posdoctoral de Paola Cristina Bello Medina dentro del programa de Becas Posdoctorales en la UNAM (POSDOC).

