





La enfermedad de Alzheimer (EA) es la primera causa de demencia y una de las principales causas de discapacidad y dependencia que afecta la calidad de vida de los adultos mayores y de sus familiares. En la actualidad el manejo farmacológico disponible incluye a los fármacos inhibidores de la acetilcolinesterasa (donepezilo, galantamina, rivastigmina) y a la memantina. Sin embargo, se ha reportado que solo un tercio de los pacientes responden al tratamiento. Se ha evidenciado que los factores genéticos pueden explicar parte de la variabilidad en la respuesta a estos fármacos.
DesarrolloEn esta revisión se incluyen los estudios farmacogenéticos de fármacos modificadores de EA, los farmacogenes analizados y los fenotipos que fueron evaluados, además de las consideraciones metodológicas que es importante tomar en cuenta en este tipo de estudios. Se encontraron 33 reportes de farmacogenética de EA en los que principalmente se ha estudiado la variabilidad en la respuesta y en el metabolismo de donepezilo. La población más estudiada es la caucásica, aunque también han sido investigados coreanos, indios y brasileños. Los biomarcadores más estudiados son CYP2D6 y APOE. Los resultados de las asociaciones son controversiales.
ConclusionesSe han identificado posibles biomarcadores farmacogenéticos para el tratamiento de EA; sin embargo, se requieren más estudios farmacogenéticos en otras poblaciones que no han sido investigadas, así como profundizar en la identificación de los biomarcadores. Este conocimiento podría ayudar a predecir la respuesta a fármacos modificadores de EA y contribuiría a tomar mejores decisiones en el tratamiento de la enfermedad en un contexto tan complejo como es el envejecimiento.
Alzheimer disease (AD) is the most common cause of dementia and is considered one of the main causes of disability and dependence affecting quality of life in elderly people and their families. Current pharmacological treatment includes acetylcholinesterase inhibitors (donepezil, galantamine, rivastigmine) and memantine; however, only one-third of patients respond to treatment. Genetic factors have been shown to play a role in this inter-individual variability in drug response.
DevelopmentWe review pharmacogenetic reports of AD-modifying drugs, the pharmacogenetic biomarkers included, and the phenotypes evaluated. We also discuss relevant methodological considerations for the design of pharmacogenetic studies into AD. A total of 33 pharmacogenetic reports were found; the majority of these focused on the variability in response to and metabolism of donepezil. Most of the patients included were from Caucasian populations, although some studies also include Korean, Indian, and Brazilian patients. CYP2D6 and APOE are the most frequently studied biomarkers. The associations proposed are controversial.
ConclusionsPotential pharmacogenetic biomarkers for AD have been identified; however, it is still necessary to conduct further research into other populations and to identify new biomarkers. This information could assist in predicting patient response to these drugs and contribute to better treatment decision-making in a context as complex as aging.
El porcentaje de adultos mayores (AM) en el mundo va en aumento constante y con ello la prevalencia de enfermedades crónico-degenerativas, entre las que se encuentran las enfermedades neurodegenerativas como las demencias1.
La demencia se caracteriza por el declinar progresivo e irreversible de funciones cognitivas (atención, orientación, memoria, lenguaje, función visoespacial, funciones ejecutivas, praxias) y de la conducta. Estas alteraciones impactan en forma progresiva y afectan la calidad de vida tanto del AM, como de la familia y el cuidador primario, y contribuyen a que las demencias sean consideradas dentro de las principales causas de discapacidad y dependencia2. Por ello, las demencias han sido señaladas como una prioridad de salud pública global por la Organización Mundial de la Salud (OMS), en 2012, y por el G8 (Grupo de los Ocho) en el 20141,3,4.
En general la prevalencia de la demencia oscila entre un 5 y un 10% de los AM de 65 años, cifra que se duplica cada 5 años hasta alcanzar una prevalencia del 25-50% en la población mayor de 85 años5.
La enfermedad de Alzheimer (EA) es la principal causa de demencia (el 60% de los casos de demencia identificados) y se define como un trastorno neurodegenerativo caracterizado por la disminución en la función neuronal que conduce a la pérdida sináptica y muerte de neuronas, lo que produce un deterioro persistente de las funciones cognitivas, altera la capacidad funcional y condiciona discapacidad y dependencia de manera gradual y progresiva6-8.
De acuerdo con el informe de la ADI (Alzheimer's Disease International) en 2015 se calculó que en el mundo había 46 millones de personas con demencia y se proyectó que aumentarían hasta 131,5 millones para el 2050. El informe destaca que casi 2/3 de las personas afectadas con algún tipo de demencia viven en países en vías de desarrollo (entre los que se incluyen países de América Latina y el Caribe) y es donde se espera que se presente un mayor incremento de casos de demencia en años venideros1. En estos países existe un menor acceso a servicios médicos especializados en comparación con los países de mayor desarrollo socioeconómico. Sin embargo, cabe resaltar que tanto en unos países como en otros, el entorno social es un factor de riesgo importante para el progreso de la enfermedad y el apoyo de los servicios sociales es fundamental para la atención al paciente con demencia.
A partir de los descubrimientos del péptido β amiloide (Aβ) y de la proteína tau (componentes principales de las placas y las marañas neurofibrilares) la investigación de la fisiopatología de EA ha proporcionado información muy importante sobre los cambios moleculares patogenéticos a nivel neuronal. Aunque la presencia de estos cambios patológicos en la EA son un sine qua non para el diagnóstico y suficientes para causar los síntomas cognitivos y conductuales en algunos pacientes (deterioro de la memoria y disfunción ejecutiva que afectan las actividades de la vida diaria), diversos factores de riesgo están asociados en pacientes que llegan a presentar síntomas después de los 75 años7,8. Sin embargo, la fisiopatología de la EA es muy compleja y el conocimiento de la enfermedad sigue siendo incompleto, por lo que ha sido insuficiente para el desarrollo de nuevos fármacos que curen, detengan o retrasen el progreso de la enfermedad de manera determinante.
La mayoría de los casos de EA se presentan después de los 65 años, son esporádicos y su origen es multifactorial; en ellos, el alelo ɛ4 del gen APOE se ha identificado como un factor de riesgo importante9. En contraste, las formas familiares son de inicio temprano y se heredan de forma autosómica dominante. Las principales mutaciones causantes de la EA familiar se han identificado en los genes que codifican para la proteína precursora de amiloide (PPA), presenilina 1 (PSEN1) y presenilina 2 (PSEN2).
Dos puntos esenciales en el manejo adecuado de la EA son el diagnóstico oportuno y temprano, y la selección del tratamiento óptimo para el paciente. El diagnóstico diferencial de la EA es complejo y requiere de pruebas tanto de imagen como neuropsicológicas, evaluaciones clínicas al paciente y entrevista con el cuidador primario o familiar. En 1984, el National Institute of Neurological and Communicative Disorders y la Alzheimer's Disease and Related Disorders Association publicaron los primeros criterios diagnósticos de la EA, nombrados NINCDS-ADRDA10. Estos fueron actualizados en 2007 con los criterios propuestos por el grupo de Dubois11 y que, a su vez, fueron revisados en 2011 por el National Institute on Aging/Alzheimer Association y nombrados como los criterios NIA-AA12.
En los casos familiares las pruebas genéticas ayudan a realizar un diagnóstico certero de la EA, que es importante para el inicio de un tratamiento en etapas tempranas de la enfermedad y para brindar un asesoramiento genético adecuado a los familiares en riesgo13,14.
Tratamientos disponibles y variabilidad en la respuestaEn la actualidad se cuenta con 2tipos de tratamientos en la EA. El tratamiento no farmacológico, que incluye entrenamiento de la memoria, estimulación social y mental, terapia musical, aromaterapia y programas de ejercicio físico, entre otros. Esta estrategia tiene el objetivo de enlentecer la progresión del deterioro cognitivo y funcional, conservar y potenciar las capacidades y habilidades (cognitivas, funcionales y sociales) preservadas, restaurar habilidades cognitivas en desuso, evitar la desconexión con el entorno y fortalecer las relaciones sociales2. Los trabajos que han evaluado la eficacia del manejo no farmacológico muestran que es una terapia coste-efectiva con resultados positivos, como demora en la institucionalización y mejoría cognitiva, que repercuten en la calidad de vida del paciente y de su cuidador15,16.
Esta revisión solo se enfoca al tratamiento farmacológico disponible, que comprende un pequeño número de fármacos utilizados en el manejo de demencias. Actualmente las agencias reguladoras internacionales solo han aprobado 3fármacos inhibidores de la acetilcolinesterasa (donepezilo, rivastigmina, galantamina) y la memantina. Sin embargo, la eficacia de estos fármacos es heterogénea y considerada de baja a media. Un estudio reciente calculó una tasa de respuesta del 27,8% a 3de estos fármacos17. Además, se han reportado diversas reacciones adversas asociadas al tratamiento de la EA como diarrea, náuseas, inestabilidad, vómitos, pérdida de peso y otras de mayor severidad como úlcera estomacal, síncope y crisis convulsivas generalizadas, por las cuales el paciente que las presenta puede requerir la suspensión del fármaco y, con ello, ve limitadas sus opciones terapéuticas18-20.
En este sentido, la farmacogenética puede ayudar a explicar cómo la variabilidad genética en los pacientes contribuye a las diferencias en la respuesta y metabolismo de los fármacos, incluyendo los modificadores de EA.
Estamos frente a la oportunidad de innovar modelos de diagnóstico y tratamiento enfocados a las demencias que tengan por objeto la preservación de la capacidad funcional y mejorar la calidad de vida de los AM. Los retos que tenemos ante nosotros son inéditos, complejos e interesantes, y suponen una nueva visión del diagnóstico y tratamiento que implica el uso de herramientas de biología molecular y farmacogenética. Por ello, es evidente la necesidad de implementar biomarcadores moleculares en el diagnóstico temprano en la EA con el objetivo de coadyuvar en el diagnóstico diferencial, en el proceso de prevención y en la atención a las personas que presentan algún tipo de demencia.
En esta revisión se describen los estudios farmacogenéticos de fármacos modificadores de EA, los farmacogenes analizados y los fenotipos que se evaluaron, además de consideraciones metodológicas importantes que tomar en cuenta en este tipo de estudios. Para ello, se realizó una investigación sistemática a febrero de 2018 en Pubmed y PharmGKB incluyendo los términos de búsqueda Alzheimer, pharmacogenetics, pharmacogenomics, donepezil, galantamine, rivastigmine y memantine.
Farmacogenética de fármacos modificadores de la enfermedad de AlzheimerSe ha reportado que entre el 75 y el 85% de la variabilidad en la respuesta terapéutica a donepezilo y a otros inhibidores de acetilcolinesterasa metabolizados por enzimas del citocromo P450 (CYP) es debida a factores farmacogenéticos, por lo que la consideración de estos factores en la terapia farmacológica de EA puede ayudar a obtener un tratamiento más eficaz y seguro en esta enfermedad21.
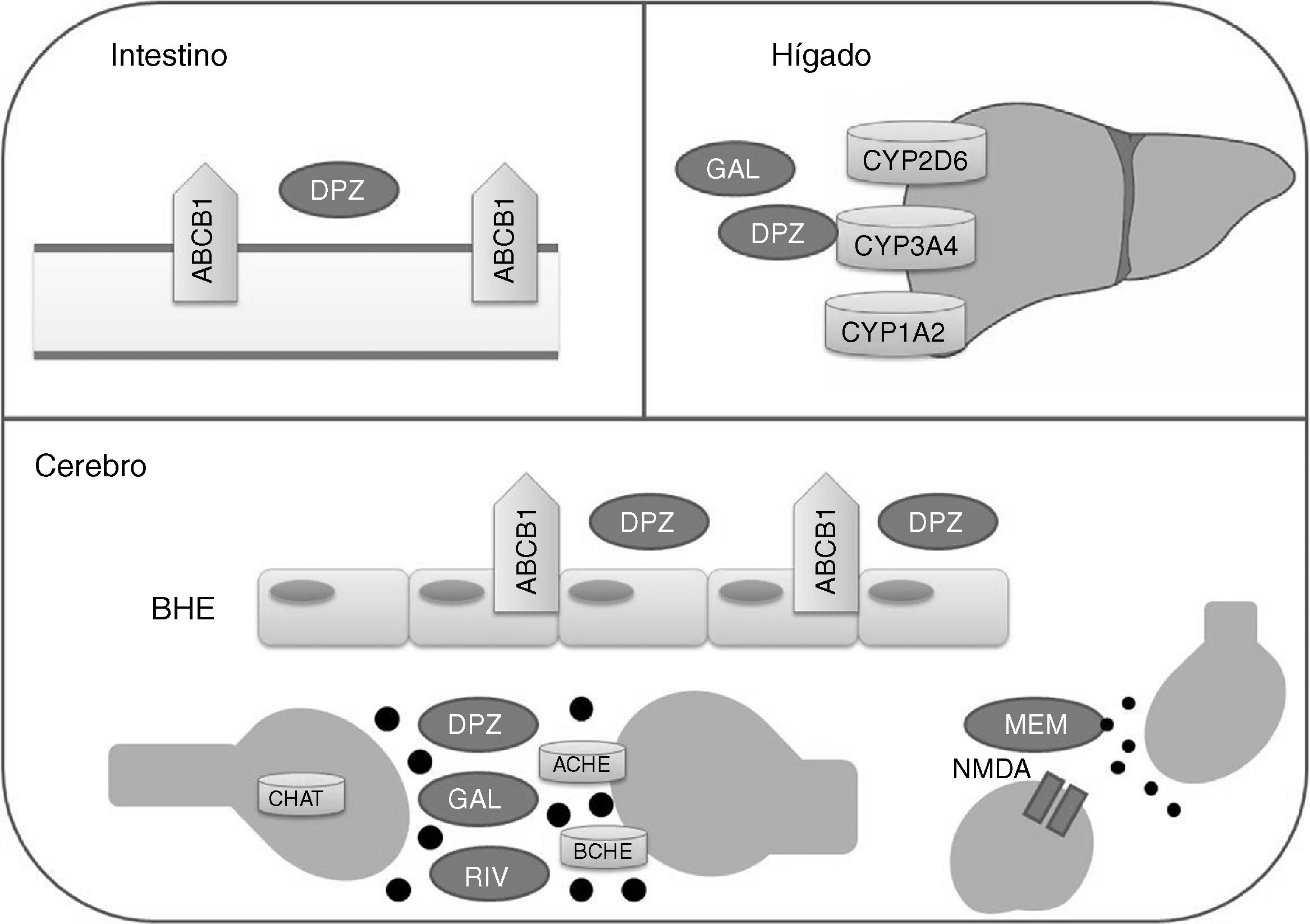
Los fármacos inhibidores de la acetilcolinesterasa y la memantina presentan rutas metabólicas distintas (fig. 1). Donepezilo y galantamina presentan un metabolismo hepático principalmente catalizado por las enzimas CYP3A4, CYP2D6 y CYP1A2, mientras que la biotransformación de rivastigmina se realiza por carbamilación. La memantina presenta un bajo metabolismo hepático y se elimina en gran parte de manera inalterada por vía renal. En cuanto a la farmacodinamia, los inhibidores de la acetilcolinesterasa presentan afinidades distintas por ACHE (acetilcolinesterasa) y BCHE (butirilcolinesterasa), donepezilo y galantamina inhiben en mayor medida a ACHE que a BCHE, mientras que rivastigmina presenta un poder inhibitorio igual para ambas enzimas21,22. ACHE es una de las enzimas más importantes en la función y respuesta nerviosa al catalizar la hidrólisis de acetilcolina tanto en el sistema nervioso autónomo como en el periférico. Por su parte, BCHE, además de acetilcolina, hidroliza butirilcolina y se encuentra ampliamente distribuida en hígado, pulmones, corazón y cerebro23. La unión de los fármacos a ACHE y BCHE consigue inhibir la hidrólisis de acetilcolina en el hipocampo para aumentar los niveles de este neurotransmisor en el espacio sináptico y que pueda unirse a los receptores colinérgicos postsinápticos. Esto contribuye a la mejora de los síntomas cognitivos, ya que se ha reportado que la neurotransmisión colinérgica, involucrada en procesos de memoria, atención y emoción, se ve seriamente afectada en la EA22.

Proteínas con las que interactúan fármacos modificadores de la enfermedad de Alzheimer.
ACHE: acetilcolinesterasa; BCHE: butirilcolinesterasa; BHE: barrera hematoencefálica; CHAT: acetiltransferasa de colina; DPZ: donepezilo; GAL: galantamina; MEM: memantina; NMDA: N-metil-D-aspartato; RIV: rivastigmina.
Por su parte, memantina presenta una moderada afinidad por el receptor NMDA (N-metil-D-aspartato) para realizar el efecto antagónico sobre él, con lo cual se inhibe el efecto patológico de niveles elevados del glutamato, que conllevan la muerte neuronal y el daño celular característicos de la EA22.
En cuanto al transporte de fármacos, se ha demostrado que la glicoproteína P tiene un papel importante en el paso de donepezilo a través de la barrera hematoencefálica24.
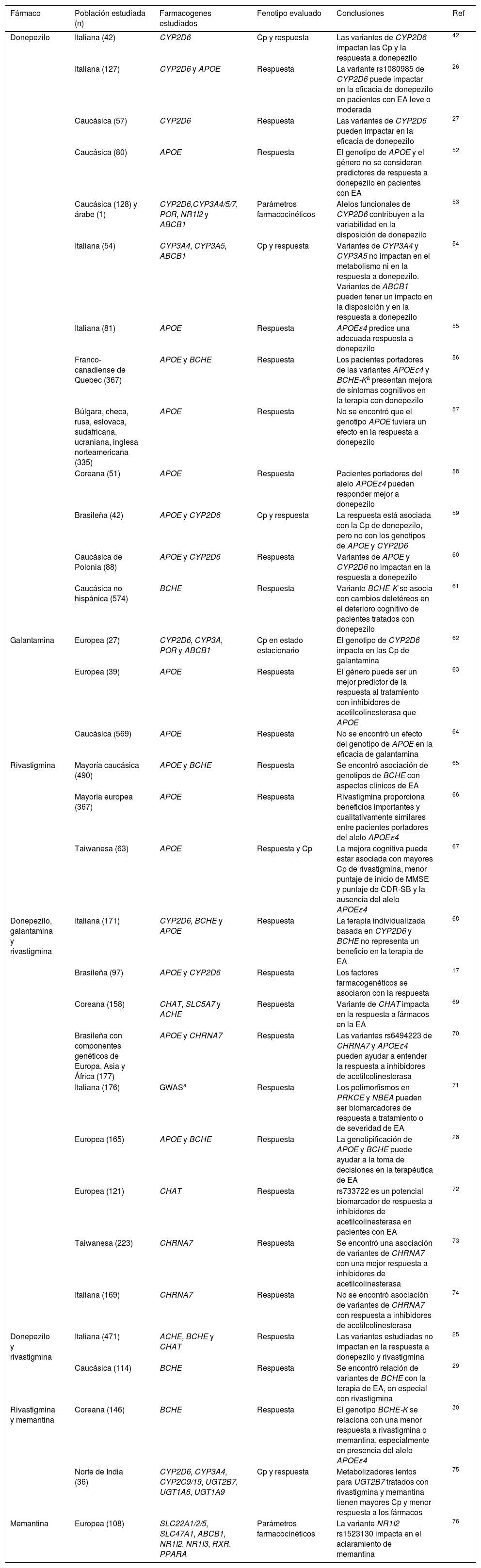
Estudios farmacogenéticos de fármacos modificadores de la enfermedad de AlzheimerEn la tabla 1 se resume la información relevante de los estudios farmacogenéticos de fármacos modificadores de EA publicados en la actualidad. Se encontraron 33 investigaciones en las que el fármaco más estudiado ha sido donepezilo (24 de los 33 reportes lo incluyen), mientras que solo 3de ellos incluyen pacientes tratados con memantina. La población más estudiada en la farmacogenética de EA es la caucásica de origen europeo, aunque también se han incluido algunos reportes en coreanos, indios y brasileños, entre otros. La mayoría de los estudios evalúan la asociación de variantes farmacogenéticas con la respuesta a fármacos modificadores de EA; sin embargo, 8de ellos también evalúan parámetros farmacocinéticos y concentraciones plasmáticas de dichos fármacos.
Estudios farmacogenéticos en fármacos modificadores de la enfermedad de Alzheimer
| Fármaco | Población estudiada (n) | Farmacogenes estudiados | Fenotipo evaluado | Conclusiones | Ref |
|---|---|---|---|---|---|
| Donepezilo | Italiana (42) | CYP2D6 | Cp y respuesta | Las variantes de CYP2D6 impactan las Cp y la respuesta a donepezilo | 42 |
| Italiana (127) | CYP2D6 y APOE | Respuesta | La variante rs1080985 de CYP2D6 puede impactar en la eficacia de donepezilo en pacientes con EA leve o moderada | 26 | |
| Caucásica (57) | CYP2D6 | Respuesta | Las variantes de CYP2D6 pueden impactar en la eficacia de donepezilo | 27 | |
| Caucásica (80) | APOE | Respuesta | El genotipo de APOE y el género no se consideran predictores de respuesta a donepezilo en pacientes con EA | 52 | |
| Caucásica (128) y árabe (1) | CYP2D6,CYP3A4/5/7, POR, NR1I2 y ABCB1 | Parámetros farmacocinéticos | Alelos funcionales de CYP2D6 contribuyen a la variabilidad en la disposición de donepezilo | 53 | |
| Italiana (54) | CYP3A4, CYP3A5, ABCB1 | Cp y respuesta | Variantes de CYP3A4 y CYP3A5 no impactan en el metabolismo ni en la respuesta a donepezilo. Variantes de ABCB1 pueden tener un impacto en la disposición y en la respuesta a donepezilo | 54 | |
| Italiana (81) | APOE | Respuesta | APOEɛ4 predice una adecuada respuesta a donepezilo | 55 | |
| Franco-canadiense de Quebec (367) | APOE y BCHE | Respuesta | Los pacientes portadores de las variantes APOEɛ4 y BCHE-Ka presentan mejora de síntomas cognitivos en la terapia con donepezilo | 56 | |
| Búlgara, checa, rusa, eslovaca, sudafricana, ucraniana, inglesa norteamericana (335) | APOE | Respuesta | No se encontró que el genotipo APOE tuviera un efecto en la respuesta a donepezilo | 57 | |
| Coreana (51) | APOE | Respuesta | Pacientes portadores del alelo APOEɛ4 pueden responder mejor a donepezilo | 58 | |
| Brasileña (42) | APOE y CYP2D6 | Cp y respuesta | La respuesta está asociada con la Cp de donepezilo, pero no con los genotipos de APOE y CYP2D6 | 59 | |
| Caucásica de Polonia (88) | APOE y CYP2D6 | Respuesta | Variantes de APOE y CYP2D6 no impactan en la respuesta a donepezilo | 60 | |
| Caucásica no hispánica (574) | BCHE | Respuesta | Variante BCHE-K se asocia con cambios deletéreos en el deterioro cognitivo de pacientes tratados con donepezilo | 61 | |
| Galantamina | Europea (27) | CYP2D6, CYP3A, POR y ABCB1 | Cp en estado estacionario | El genotipo de CYP2D6 impacta en las Cp de galantamina | 62 |
| Europea (39) | APOE | Respuesta | El género puede ser un mejor predictor de la respuesta al tratamiento con inhibidores de acetilcolinesterasa que APOE | 63 | |
| Caucásica (569) | APOE | Respuesta | No se encontró un efecto del genotipo de APOE en la eficacia de galantamina | 64 | |
| Rivastigmina | Mayoría caucásica (490) | APOE y BCHE | Respuesta | Se encontró asociación de genotipos de BCHE con aspectos clínicos de EA | 65 |
| Mayoría europea (367) | APOE | Respuesta | Rivastigmina proporciona beneficios importantes y cualitativamente similares entre pacientes portadores del alelo APOEɛ4 | 66 | |
| Taiwanesa (63) | APOE | Respuesta y Cp | La mejora cognitiva puede estar asociada con mayores Cp de rivastigmina, menor puntaje de inicio de MMSE y puntaje de CDR-SB y la ausencia del alelo APOEɛ4 | 67 | |
| Donepezilo, galantamina y rivastigmina | Italiana (171) | CYP2D6, BCHE y APOE | Respuesta | La terapia individualizada basada en CYP2D6 y BCHE no representa un beneficio en la terapia de EA | 68 |
| Brasileña (97) | APOE y CYP2D6 | Respuesta | Los factores farmacogenéticos se asociaron con la respuesta | 17 | |
| Coreana (158) | CHAT, SLC5A7 y ACHE | Respuesta | Variante de CHAT impacta en la respuesta a fármacos en la EA | 69 | |
| Brasileña con componentes genéticos de Europa, Asia y África (177) | APOE y CHRNA7 | Respuesta | Las variantes rs6494223 de CHRNA7 y APOEɛ4 pueden ayudar a entender la respuesta a inhibidores de acetilcolinesterasa | 70 | |
| Italiana (176) | GWASa | Respuesta | Los polimorfismos en PRKCE y NBEA pueden ser biomarcadores de respuesta a tratamiento o de severidad de EA | 71 | |
| Europea (165) | APOE y BCHE | Respuesta | La genotipificación de APOE y BCHE puede ayudar a la toma de decisiones en la terapéutica de EA | 28 | |
| Europea (121) | CHAT | Respuesta | rs733722 es un potencial biomarcador de respuesta a inhibidores de acetilcolinesterasa en pacientes con EA | 72 | |
| Taiwanesa (223) | CHRNA7 | Respuesta | Se encontró una asociación de variantes de CHRNA7 con una mejor respuesta a inhibidores de acetilcolinesterasa | 73 | |
| Italiana (169) | CHRNA7 | Respuesta | No se encontró asociación de variantes de CHRNA7 con respuesta a inhibidores de acetilcolinesterasa | 74 | |
| Donepezilo y rivastigmina | Italiana (471) | ACHE, BCHE y CHAT | Respuesta | Las variantes estudiadas no impactan en la respuesta a donepezilo y rivastigmina | 25 |
| Caucásica (114) | BCHE | Respuesta | Se encontró relación de variantes de BCHE con la terapia de EA, en especial con rivastigmina | 29 | |
| Rivastigmina y memantina | Coreana (146) | BCHE | Respuesta | El genotipo BCHE-K se relaciona con una menor respuesta a rivastigmina o memantina, especialmente en presencia del alelo APOEɛ4 | 30 |
| Norte de India (36) | CYP2D6, CYP3A4, CYP2C9/19, UGT2B7, UGT1A6, UGT1A9 | Cp y respuesta | Metabolizadores lentos para UGT2B7 tratados con rivastigmina y memantina tienen mayores Cp y menor respuesta a los fármacos | 75 | |
| Memantina | Europea (108) | SLC22A1/2/5, SLC47A1, ABCB1, NR1I2, NR1I3, RXR, PPARA | Parámetros farmacocinéticos | La variante NR1I2 rs1523130 impacta en el aclaramiento de memantina | 76 |
En este trabajo no se evaluó la asociación de respuesta con un farmacogén en específico sino que se realizó un estudio del genoma completo en pacientes respondedores y no respondedores al tratamiento con inhibidores de acetilcolinesterasa.
CDR-SB: escala de valoración de demencia clínica, suma de cajas; Cp: concentraciones plasmáticas; EA: enfermedad de Alzheimer; GWAS: por sus siglas en inglés, Genome-Wide Association Study; MMSE: por sus siglas en inglés, Mini Mental State Examination; n: número de pacientes incluidos en el estudio; Ref: referencia.
La mayor parte de las asociaciones positivas reportadas han sido entre las variantes de CYP2D6 con concentraciones plasmáticas y la respuesta a fármacos inhibidores de acetilcolinesterasa; mientras que los resultados obtenidos en los estudios de asociación de respuesta terapéutica y variantes de APOE y BCHE han sido controversiales. A pesar de que existen pocos estudios en los que se ha incluido a pacientes tratados con memantina, las variantes de UGT2B7, BCHE y NR1I2 se han encontrado relacionadas con parámetros farmacocinéticos y con la variación en la respuesta a este fármaco (tabla 1).
Entre los farmacogenes considerados en los estudios se encuentran: ABCB1, ACHE, APOE, BCHE, CHAT, CHRNA7, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5, CYP3A7, NR1I2, NR1I3, POR, PPAR, RXR, SLC22A1/2/5, SLC47A1, SLC5A7, UGT1A6, UGT1A9 y UGT2B7. La información de estos genes se describe en la tabla 2. El gen APOE ha sido estudiado en mayor medida con el riesgo para desarrollar EA y se ha reportado que los portadores del alelo ɛ4 presentan un mayor riesgo en comparación con los que presentan el alelo ɛ3, mientras que el alelo ɛ2 se ha identificado como un factor de protección9. Por su parte, los genes ACHE, BCHE, CHAT, CHRNA7 y SLC5A7 participan directamente en la formación y el metabolismo de la acetilcolina25. Otros genes incluidos en los estudios farmacogenéticos están relacionados con el transporte, metabolismo e inhibición de los fármacos inhibidores de la acetilcolinesterasa (por ejemplo, ABCB1, CYP2D6, CYP3A4, CYP3A5 y CYP3A7) o con la transcripción y expresión de estos genes (POR, NR1I2, NR1I3, RXR, PPAR, entre otros).
Genes incluidos en los estudios farmacogenéticos de fármacos modificadores de demencia
| Símbolo | Localización | Descripcióna |
|---|---|---|
| ABCB1 | 7q21.12 | Miembro 1 de la subfamilia B de los transportadores de unión a ATP. Es un transportador de eflujo, también conocido como glicoproteína-P, que transporta xenobióticos fuera de la célula |
| ACHE | 7q22 | Acetilcolinesterasa. Se encarga de la hidrólisis de acetilcolina en la sinapsis de neuronas colinérgicas para finalizar la señalización |
| APOE | 19q13.2 | Apolipoproteína E. Se une a un receptor celular específico ubicado en el hígado y tejido periférico, y es esencial para el catabolismo normal de lipoproteínas ricas en triglicéridos |
| BCHE | 3q26.1-q26.2 | Butirilcolinesterasa. Es un tipo de colinesterasa y miembro de la familia de las proteínas carboxilesterasa tipo B/lipasa. Participa en la desintoxicación de sustancias tóxicas como compuestos organofosforados y en el metabolismo de sustancias como cocaína, heroína y aspirina |
| CHAT | 10q11.2 | Acetiltransferasa de colina. Enzima que cataliza la biosíntesis de acetilcolina |
| CHRNA7 | 15q14 | Subunidad alfa 7 del receptor nicotínico de acetilcolina. Este tipo de receptores pertenecen a una superfamilia de canales iónicos dependientes de ligando que participa en la transmisión de señales en la sinapsis |
| CYP2C9 | 10q24 | Miembro 9 subfamilia C familia 2 del citocromo P450. Monooxigenasa que cataliza diversas reacciones del metabolismo de fármacos (fenitoína, tolbutamida, ibuprofeno y S-warfarina) y síntesis de colesterol, esteroides y otros lípidos |
| CYP2C19 | 10q24 | Miembro 19 subfamilia C familia 2 del citocromo P450. Monooxigenasa que participa en la síntesis de colesterol, esteroides y otros lípidos y en el metabolismo de omeprazol, diazepam y algunos barbitúricos |
| CYP2D6 | 22q13.1 | Miembro 6 subfamilia D familia 2 del citocromo P450. Monooxigenasa que participa en la síntesis de colesterol, esteroides y otros lípidos y en el metabolismo de antidepresivos, antipsicóticos, analgésicos, antitusivos, bloqueadores betaadrenérgicos, antiarrítmicos, antieméticos |
| CYP3A4 | 7q21.1 | Miembro 4 subfamilia A familia 3 del citocromo P450. Monooxigenasa que participa en el metabolismo de esteroides y sustancias cancerígenas, y aproximadamente en la mitad de los fármacos de uso común como acetaminofén, codeína, ciclosporina A, diazepam y eritromicina |
| CYP3A5 | 7q21.1 | Miembro 5 subfamilia A familia 3 del citocromo P450. Monooxigenasa que participa en el metabolismo de fármacos y de hormonas como la testosterona y la progesterona |
| CYP3A7 | 7q22.1 | Miembro 7 subfamilia A familia 3 del citocromo P450. Monooxigenasa que hidroliza testosterona y 3-sulfato dehidroepiandrosterona, que participa en la formación de estriol durante el embarazo |
| NR1I2 | 3q12-q13.3 | Miembro 2 grupo I subfamilia 1 de receptores nucleares. Factores de transcripción caracterizados por tener un dominio de unión a ligando y un dominio de unión a ADN. Es un regulador de la transcripción de CYP3A4 |
| NR1I3 | 1q23.3 | Miembro 3 grupo I subfamilia 1 de receptores nucleares. Es un regulador del metabolismo de compuestos xenobióticos y endobióticos. Regula la transcripción de genes involucrados en el metabolismo de fármacos y la eliminación de bilirrubina, como por ejemplo los miembros de la familia del citocromo P450 |
| POR | 7q11.2 | Citocromo P450 oxidorreductasa. Oxidorreductadasa que se une a 2 cofactores, FAD y FMN, que donan electrones desde NADPH a todas las enzimas microsomales P450 |
| PPARA | 22q13.31 | Receptor alfa activado por proliferador de peroxisomas. Afecta la expresión de genes involucrados en la proliferación celular, diferenciación celular y en las respuestas inflamatoria e inmunológica |
| RXR | 9q34.3 | Receptor X retinoide. Receptor nuclear que interviene en los efectos biológicos de los retinoides por su participación en la activación de genes mediada por ácido retinoico |
| SLC22A1, SLC22A2 y SLC22A5 | 6q25.3 | Miembros 1, 2 y 5, familia 22 de los transportadores de soluto. Participan de manera importante en la eliminación de cationes orgánicos pequeños endógenos así como fármacos y sustancias tóxicas ambientales |
| SLC47A1 | 17p11.2 | Miembro 1 familia 47 de los transportadores de soluto. Proteína transportadora de función desconocida |
| SLC5A7 | 2q12 | Miembro 7 familia 5 de los transportadores de soluto. Transportador de alta afinidad dependiente de sodio y cloro que media la captura de colina para la síntesis de acetilcolina en neuronas colinérgicas |
| UGT1A6 y UGT1A9 | 2q37 | Miembros A6 y A9 familia 1 de UDP glucuronosiltransferasa. Participan en la glucuronidación de moléculas pequeñas lipofílicas como esteroides, bilirrubina, hormonas y fármacos para convertirlas en metabolitos hidrofílicos |
| UGT2B7 | 4q13 | Miembro B7 familia 2 de UDP glucuronosiltransferasa. Participa en la conjugación y eliminación de compuestos endógenos y xenobióticos. Presenta afinidad específica por estriol y 3,4-catecol estrógenos |
ADN: ácido desoxirribonucleico; ATP: adenosina trifosfato; FAD: flavina adenina dinucleótido; FMN: flavina mononucleótido; NADPH: nicotinamida adenina dinucleótido fosfato; UDP: uridina difosfato.
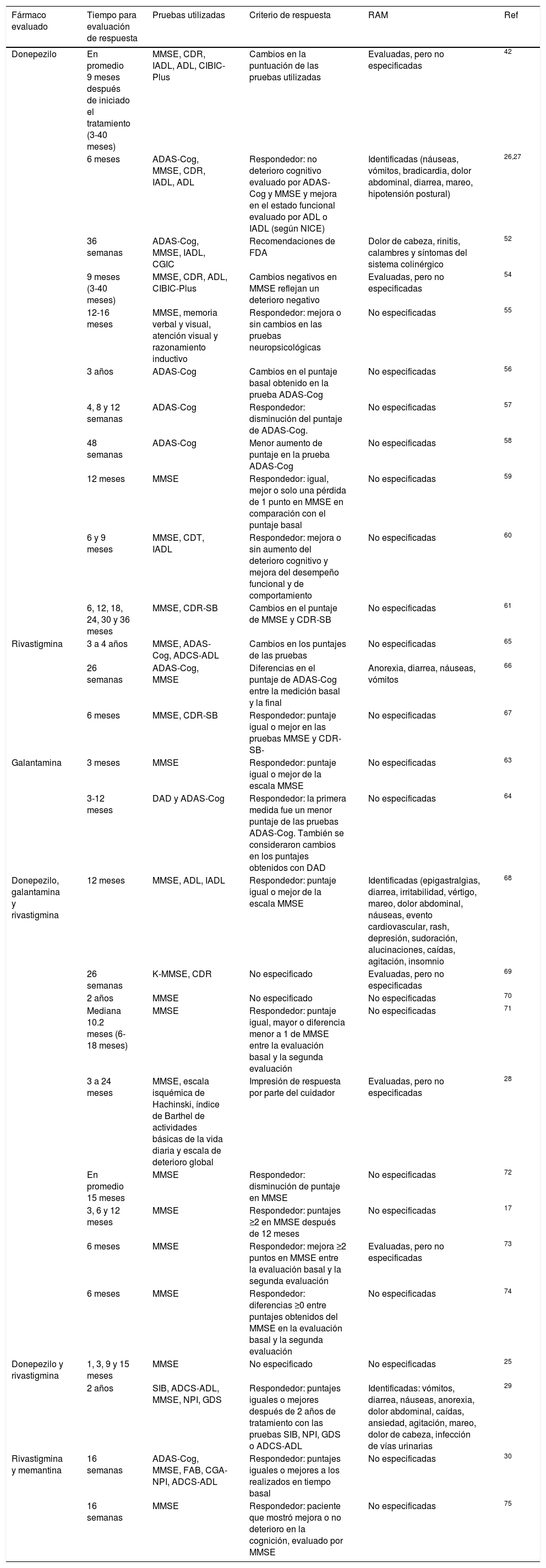
Para estudiar la asociación de las variantes genéticas con la respuesta terapéutica a fármacos modificadores de EA se requiere de parámetros clínicos establecidos y de pruebas neuropsicológicas que evalúen la cognición después de iniciado el tratamiento. Los estudios farmacogenéticos encontrados en la literatura utilizan los requerimientos establecidos por el Instituto Nacional de Salud y Excelencia Clínica (NICE, por sus siglas en inglés National Institute for Health and Clinical Excellence) y por la Administración de Fármacos y Alimentos de Estados Unidos (FDA, por sus siglas en inglés Food and Drug Administration).
Algunos estudios que se basaron en las recomendaciones de NICE consideraron respondedores a aquellos que no presentaran deterioro cognitivo después de, al menos, 6 meses de tratamiento. Esta respuesta fue evaluada por un mejor o igual puntaje de las pruebas Mini-mental de Fostein (MMSE por sus siglas en inglés Mini Mental State Examination) y ADAS-Cog (por sus siglas en inglés Alzheimer's Disease Assessment Scale – Cognitive section) en comparación con el puntaje basal y una mejora en el estado funcional determinado por la valoración de actividades de la vida diaria o índice de Katz (ADL, por sus siglas en inglés activities of daily living) o por la prueba de Lawton-Brody de actividades instrumentales de la vida diaria (IADL, por sus siglas en inglés instrumental activities of daily living)26,27. La mayoría de los estudios se enfocaron en el puntaje arrojado por MMSE, aunque otros reportes incluyeron más pruebas para determinar la respuesta a fármacos modificadores de EA28-30 (tabla 3).
Aspectos relevantes para evaluación de respuesta clínica a fármacos modificadores de enfermedad de Alzheimer utilizados en los estudios farmacogenéticos
| Fármaco evaluado | Tiempo para evaluación de respuesta | Pruebas utilizadas | Criterio de respuesta | RAM | Ref |
|---|---|---|---|---|---|
| Donepezilo | En promedio 9 meses después de iniciado el tratamiento (3-40 meses) | MMSE, CDR, IADL, ADL, CIBIC-Plus | Cambios en la puntuación de las pruebas utilizadas | Evaluadas, pero no especificadas | 42 |
| 6 meses | ADAS-Cog, MMSE, CDR, IADL, ADL | Respondedor: no deterioro cognitivo evaluado por ADAS-Cog y MMSE y mejora en el estado funcional evaluado por ADL o IADL (según NICE) | Identificadas (náuseas, vómitos, bradicardia, dolor abdominal, diarrea, mareo, hipotensión postural) | 26,27 | |
| 36 semanas | ADAS-Cog, MMSE, IADL, CGIC | Recomendaciones de FDA | Dolor de cabeza, rinitis, calambres y síntomas del sistema colinérgico | 52 | |
| 9 meses (3-40 meses) | MMSE, CDR, ADL, CIBIC-Plus | Cambios negativos en MMSE reflejan un deterioro negativo | Evaluadas, pero no especificadas | 54 | |
| 12-16 meses | MMSE, memoria verbal y visual, atención visual y razonamiento inductivo | Respondedor: mejora o sin cambios en las pruebas neuropsicológicas | No especificadas | 55 | |
| 3 años | ADAS-Cog | Cambios en el puntaje basal obtenido en la prueba ADAS-Cog | No especificadas | 56 | |
| 4, 8 y 12 semanas | ADAS-Cog | Respondedor: disminución del puntaje de ADAS-Cog. | No especificadas | 57 | |
| 48 semanas | ADAS-Cog | Menor aumento de puntaje en la prueba ADAS-Cog | No especificadas | 58 | |
| 12 meses | MMSE | Respondedor: igual, mejor o solo una pérdida de 1 punto en MMSE en comparación con el puntaje basal | No especificadas | 59 | |
| 6 y 9 meses | MMSE, CDT, IADL | Respondedor: mejora o sin aumento del deterioro cognitivo y mejora del desempeño funcional y de comportamiento | No especificadas | 60 | |
| 6, 12, 18, 24, 30 y 36 meses | MMSE, CDR-SB | Cambios en el puntaje de MMSE y CDR-SB | No especificadas | 61 | |
| Rivastigmina | 3 a 4 años | MMSE, ADAS-Cog, ADCS-ADL | Cambios en los puntajes de las pruebas | No especificadas | 65 |
| 26 semanas | ADAS-Cog, MMSE | Diferencias en el puntaje de ADAS-Cog entre la medición basal y la final | Anorexia, diarrea, náuseas, vómitos | 66 | |
| 6 meses | MMSE, CDR-SB | Respondedor: puntaje igual o mejor en las pruebas MMSE y CDR-SB- | No especificadas | 67 | |
| Galantamina | 3 meses | MMSE | Respondedor: puntaje igual o mejor de la escala MMSE | No especificadas | 63 |
| 3-12 meses | DAD y ADAS-Cog | Respondedor: la primera medida fue un menor puntaje de las pruebas ADAS-Cog. También se consideraron cambios en los puntajes obtenidos con DAD | No especificadas | 64 | |
| Donepezilo, galantamina y rivastigmina | 12 meses | MMSE, ADL, IADL | Respondedor: puntaje igual o mejor de la escala MMSE | Identificadas (epigastralgias, diarrea, irritabilidad, vértigo, mareo, dolor abdominal, náuseas, evento cardiovascular, rash, depresión, sudoración, alucinaciones, caídas, agitación, insomnio | 68 |
| 26 semanas | K-MMSE, CDR | No especificado | Evaluadas, pero no especificadas | 69 | |
| 2 años | MMSE | No especificado | No especificadas | 70 | |
| Mediana 10.2 meses (6-18 meses) | MMSE | Respondedor: puntaje igual, mayor o diferencia menor a 1 de MMSE entre la evaluación basal y la segunda evaluación | No especificadas | 71 | |
| 3 a 24 meses | MMSE, escala isquémica de Hachinski, índice de Barthel de actividades básicas de la vida diaria y escala de deterioro global | Impresión de respuesta por parte del cuidador | Evaluadas, pero no especificadas | 28 | |
| En promedio 15 meses | MMSE | Respondedor: disminución de puntaje en MMSE | No especificadas | 72 | |
| 3, 6 y 12 meses | MMSE | Respondedor: puntajes ≥2 en MMSE después de 12 meses | No especificadas | 17 | |
| 6 meses | MMSE | Respondedor: mejora ≥2 puntos en MMSE entre la evaluación basal y la segunda evaluación | Evaluadas, pero no especificadas | 73 | |
| 6 meses | MMSE | Respondedor: diferencias ≥0 entre puntajes obtenidos del MMSE en la evaluación basal y la segunda evaluación | No especificadas | 74 | |
| Donepezilo y rivastigmina | 1, 3, 9 y 15 meses | MMSE | No especificado | No especificadas | 25 |
| 2 años | SIB, ADCS-ADL, MMSE, NPI, GDS | Respondedor: puntajes iguales o mejores después de 2 años de tratamiento con las pruebas SIB, NPI, GDS o ADCS-ADL | Identificadas: vómitos, diarrea, náuseas, anorexia, dolor abdominal, caídas, ansiedad, agitación, mareo, dolor de cabeza, infección de vías urinarias | 29 | |
| Rivastigmina y memantina | 16 semanas | ADAS-Cog, MMSE, FAB, CGA-NPI, ADCS-ADL | Respondedor: puntajes iguales o mejores a los realizados en tiempo basal | No especificadas | 30 |
| 16 semanas | MMSE | Respondedor: paciente que mostró mejora o no deterioro en la cognición, evaluado por MMSE | No especificadas | 75 |
ADAS-Cog: por sus siglas en inglés, Alzheimer's Disease Assessment Scale-Cognitive section; ADCS-ADL: por sus siglas en inglés, Alzheimer's Disease Cooperative Study Activities of Daily Living; ADL: por sus siglas en inglés Activities of Daily Living; CDR-SB: por sus siglas en inglés, Clinical Dementia Rating sum of boxes; CDR: por sus siglas en inglés Clinical Dementia Rating; CDT: por sus siglas en inglés, Clock Drawing Test; CGA-NPI: por sus siglas en inglés, Caregiver-Administered NeuropsychiatricInventory; CIBIC-Plus: por sus siglas en inglés Clinician's Interview-Based Impression of Change Plus caregiver input; FAB: por sus siglas en inglés, Frontal Assessment Battery; FDA: por sus siglas en inglés, Food and Drug Administration; GDS: por sus siglas en inglés, Global Deterioration Scale; IADL: por sus siglas en inglés, Instrumental Activities of Daily Living; K-MMSE: MMSE versión coreana; MMSE: por sus siglas en inglés, Mini Mental State Examination; NICE: por sus siglas en inglés, National Institute for Health and Care Excellence; NPI: por sus siglas en inglés, Neuropsychiatric Inventory; RAM: reacción adversa a medicamentos; SIB: por sus siglas en inglés, Severe Impairment Battery.
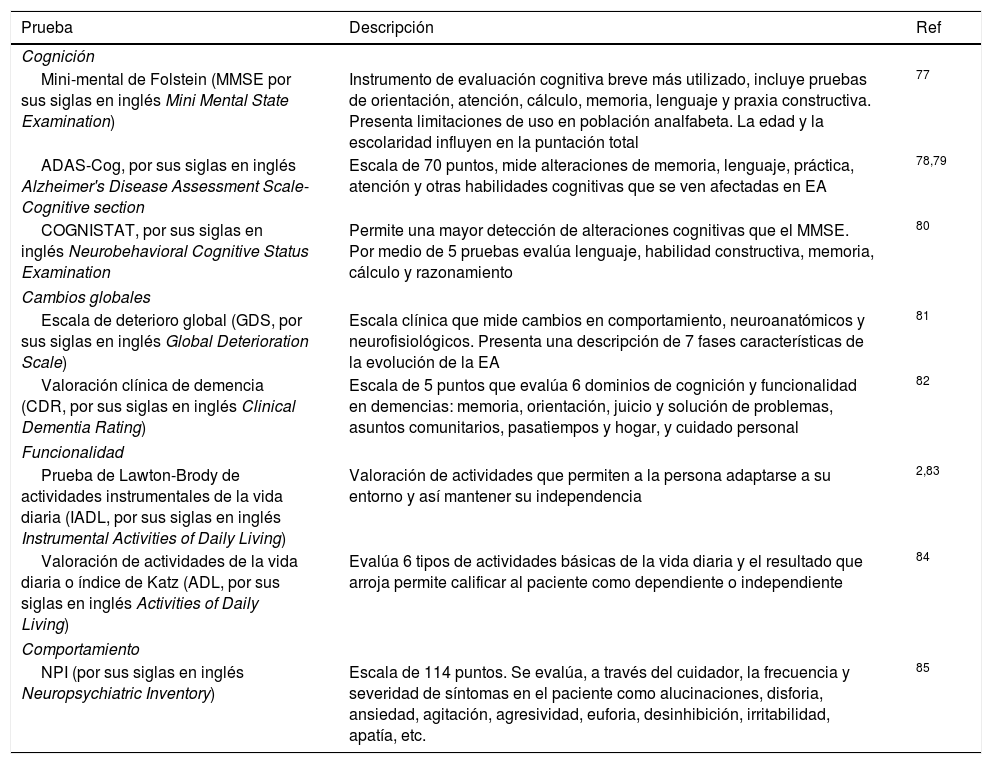
Existen diferentes tipos de pruebas de clinimetría que permiten evaluar la cognición y la funcionalidad2,31: en la tabla 4 se describen las principales pruebas utilizadas en los estudios farmacogenéticos de EA reportados. Al realizar las pruebas de funcionamiento cognitivo, es importante considerar los siguientes puntos para evitar que el estudio se vea afectado por un sesgo en los resultados32:
- -
Cuando se utilicen escalas para determinar la severidad de EA, el profesional de la salud debe tomar en cuenta cualquier discapacidad de aprendizaje, tanto sensorial como físico.
- -
La evaluación del estado cognitivo no puede basarse solo en el puntaje obtenido en pacientes que presenten alguna dificultad de comunicación lingüística o que hablen otro idioma, con un bajo nivel educativo o con alguna discapacidad.
- -
Para evaluar la severidad de la enfermedad y la respuesta a fármacos modificadores no es apropiado considerar solo el puntaje de MMSE y se debe complementar con otras pruebas para una valoración multidominio.
Principales pruebas utilizadas para evaluación de deterioro cognitivo y funcionalidad en los estudios farmacogenéticos de enfermedad de Alzheimer
| Prueba | Descripción | Ref |
|---|---|---|
| Cognición | ||
| Mini-mental de Folstein (MMSE por sus siglas en inglés Mini Mental State Examination) | Instrumento de evaluación cognitiva breve más utilizado, incluye pruebas de orientación, atención, cálculo, memoria, lenguaje y praxia constructiva. Presenta limitaciones de uso en población analfabeta. La edad y la escolaridad influyen en la puntación total | 77 |
| ADAS-Cog, por sus siglas en inglés Alzheimer's Disease Assessment Scale-Cognitive section | Escala de 70 puntos, mide alteraciones de memoria, lenguaje, práctica, atención y otras habilidades cognitivas que se ven afectadas en EA | 78,79 |
| COGNISTAT, por sus siglas en inglés Neurobehavioral Cognitive Status Examination | Permite una mayor detección de alteraciones cognitivas que el MMSE. Por medio de 5 pruebas evalúa lenguaje, habilidad constructiva, memoria, cálculo y razonamiento | 80 |
| Cambios globales | ||
| Escala de deterioro global (GDS, por sus siglas en inglés Global Deterioration Scale) | Escala clínica que mide cambios en comportamiento, neuroanatómicos y neurofisiológicos. Presenta una descripción de 7 fases características de la evolución de la EA | 81 |
| Valoración clínica de demencia (CDR, por sus siglas en inglés Clinical Dementia Rating) | Escala de 5 puntos que evalúa 6 dominios de cognición y funcionalidad en demencias: memoria, orientación, juicio y solución de problemas, asuntos comunitarios, pasatiempos y hogar, y cuidado personal | 82 |
| Funcionalidad | ||
| Prueba de Lawton-Brody de actividades instrumentales de la vida diaria (IADL, por sus siglas en inglés Instrumental Activities of Daily Living) | Valoración de actividades que permiten a la persona adaptarse a su entorno y así mantener su independencia | 2,83 |
| Valoración de actividades de la vida diaria o índice de Katz (ADL, por sus siglas en inglés Activities of Daily Living) | Evalúa 6 tipos de actividades básicas de la vida diaria y el resultado que arroja permite calificar al paciente como dependiente o independiente | 84 |
| Comportamiento | ||
| NPI (por sus siglas en inglés Neuropsychiatric Inventory) | Escala de 114 puntos. Se evalúa, a través del cuidador, la frecuencia y severidad de síntomas en el paciente como alucinaciones, disforia, ansiedad, agitación, agresividad, euforia, desinhibición, irritabilidad, apatía, etc. | 85 |
EA: enfermedad de Alzheimer.
En 8de los 33 estudios farmacogenéticos encontrados se determinaron las concentraciones plasmáticas de los fármacos modificadores de EA para realizar el estudio de asociación con las variantes genéticas seleccionadas. Estos estudios han incluido fármacos inhibidores de acetilcolinesterasa y memantina. Los aspectos relevantes sobre la metodología utilizada en estos artículos se describen en la tabla 5.
Especificaciones de los métodos utilizados para la determinación de concentraciones plasmáticas de los fármacos modificadores de enfermedad de Alzheimer en los estudios farmacogenéticos reportados
| Fármaco evaluado | Tiempo para recolección de muestra después de última administración del fármaco | Tipo de muestra | Método analítico utilizado | Referencia |
|---|---|---|---|---|
| Donepezilo | 12-15 hs | Sangre | HPLC-UV | 42,54 |
| En promedio 14 h (1-28 h) | Sangre | HPLC-MS | 53 | |
| No especificado | Sangre | HPLC-ESI-MS/MS | 59 | |
| Galantamina | 1-7 h | Sangre | HPLC-MS | 62 |
| Rivastigmina | No especificado | Sangre | MEKC-UV | 67 |
| Memantina | 0-24 h | Sangre | HPLC-MS | 76 |
| Rivastigmina y memantina | No especificado | Sangre | LC-MS/MS | 75 |
HPLC-ESI-MS/MS: cromatografía de líquidos acoplada a espectrometría de masas en tándem con ionización por electrospray; HPLC-MS: cromatografía de líquidos de alta resolución acoplado a espectrómetro de masas; HPLC-UV: cromatografía de líquidos de alta resolución con detector de ultravioleta-visible; LC-MS/MS: cromatografía de líquidos acoplada a espectrometría de masas en tándem; MEKC-UV: cromatografía electrocinética micelar con detector UV.
Las determinaciones de concentraciones plasmáticas de fármacos modificadores de EA en pacientes se realizaron en condiciones de estado estacionario, el cual se alcanza entre las 4-5vidas medias del fármaco33. La vida media que se ha reportado para donepezilo es considerablemente mayor (70 a 80 h) que para los otros inhibidores de acetilcolinesterasa (0,3 a 12 h)34-36. Por su parte, para memantina también se ha reportado una vida media larga de 60-80 h37. Debido a estos valores de vida media se puede considerar que como máximo en 17 días se alcanza el estado estacionario en la administración de estos fármacos.
La cromatografía de líquidos con detección de masas ha sido la metodología de elección para la cuantificación de fármacos modificadores de demencia en los estudios farmacogenéticos reportados. Según la literatura, este método analítico presenta ventajas como sensibilidad, especificidad, rapidez, obtención de resultados con mínimos volúmenes de plasma (250-500μL), además de que, en algunos casos, se puede cuantificar más de un fármaco de manera simultánea, o también alguno de sus metabolitos38-41.
Aspectos importantes que considerar en el estudio farmacogenético de la enfermedad de AlzheimerDebido a la complejidad de la EA, los estudios farmacogenéticos incluyen evaluación de la salud física, la presencia de otras enfermedades y el tratamiento concomitante42, así como la adherencia al tratamiento26,27. Sin duda, estos factores pueden impactar de manera importante en la respuesta y en las concentraciones plasmáticas de los fármacos modificadores de EA y, por tanto, interferir en los estudios de asociación de estos parámetros con variantes farmacogenéticas.
Los estudios en los que se evalúa la adherencia a la terapia farmacológica en pacientes con EA reportan cifras entre el 34 y el 94% de cumplimiento en el régimen terapéutico, diferencias que pueden ser notables debido al uso de técnicas de apoyo para cumplir el tratamiento así como a la presencia de un cuidador primario que se encargue de proveer los medicamentos43. Esta variabilidad puede implicar un sesgo en la medición de respuesta realizada en los estudios farmacogenéticos, por lo que existen diversos métodos que permiten evaluar la adherencia al tratamiento44 y su utilización también contribuiría a obtener resultados más confiables en la asociación de la respuesta con variantes genéticas.
Varias investigaciones han reportado que aproximadamente el 50% de la población de AM estudiada presenta una o más enfermedades crónicas45,46. Esto puede impactar en la evaluación de la respuesta a fármacos modificadores de EA, ya que se ha reportado que la comorbilidad puede complicar el curso clínico de EA, por ejemplo, acelerando el deterioro cognitivo y la pérdida de funcionalidad. Entre los padecimientos que pueden afectar la función cognitiva de los pacientes con demencia se encuentran: insuficiencia cardiaca, enfermedad arterial coronaria, hipertensión, diabetes y enfermedad pulmonar obstructiva crónica45,47.
Se ha calculado que un paciente con demencia en atención primaria presenta en promedio 2,4 padecimientos crónicos y es tratado con 5,1 medicamentos distintos48. La politerapia puede impactar en gran medida la respuesta y las concentraciones de los fármacos modificadores de EA, sobre todo en aquellos que son metabolizados por citocromos, los cuales pueden ser fácilmente inhibidos o inducidos por otros fármacos. Sin embargo, se ha reportado que los inhibidores de acetilcolinesterasa y el antagonista del receptor NMDA (memantina) presentan un bajo potencial de interacciones farmacológicas49. A pesar de ello, el registro del tratamiento concomitante puede ser relevante en los estudios farmacogenéticos de EA, puesto que puede explicar alguna variación en la respuesta, así como la presencia de reacciones adversas45.
Inmunoterapia con anticuerpos monoclonalesActualmente, los nuevos enfoques de terapias contra la EA se basan en la hipótesis de liberación y producción del péptido Aβ y consisten en el uso de anticuerpos monoclonales que reconocen diversos epítopos del péptido Aβ y muestran una selectividad de unión diferente50. Los ensayos clínicos de bapineuzumab, ponezumab, solanezumab y gantenerumab han sido suspendidos, mientras que aducanumab se encuentra en fase III. Este anticuerpo humano está dirigido selectivamente contra los agregados de péptido Aβ (incluyendo oligómeros solubles y fibrillas insolubles), y da como resultado la reducción de las placas de Aβ, acompañada de una progresión clínica lenta en pacientes con EA prodrómica o leve. En el ensayo de fase Ib del uso de aducanumab, realizado en 196 pacientes con EA, fue importante la selección del paciente en el cual debía ser confirmada la enfermedad del depósito del Aβ mediante tomografía de emisión de positrones molecular. El anticuerpo fue administrado con seguridad y tolerabilidad aceptables, pero tuvo un efecto secundario asociado con la eliminación del Aβ, que fue dependiente de la dosis y se produjo con mayor frecuencia en portadores del alelo ɛ4 del gen APOE en comparación con los no portadores. Sin embargo, este ensayo no demostró tener la potencia necesaria para los criterios clínicos exploratorios, por lo que se recomienda que los resultados cognitivos sean interpretados con precaución51.
ConclusionesEsta revisión de la literatura sobre estudios farmacogenéticos en EA muestra que existen pocos estudios a nivel mundial en este ámbito. Sin embargo, algunos resultados muestran una asociación de distintas variantes genéticas como CYP2D6, ABCB1, BCHE, entre otros, con la respuesta o concentraciones plasmáticas de fármacos modificadores de EA.
La realización de este tipo de estudios en poblaciones que aún no han sido estudiadas puede aportar mayor información que ayude a las agencias reguladoras a formular recomendaciones farmacogenéticas más precisas en la terapéutica de EA. Por ejemplo, CYP2D6 es un biomarcador farmacogenético recomendado en la terapia de múltiples fármacos y en EA puede resultar de ayuda para el uso seguro y eficaz de fármacos inhibidores de acetilcolinesterasa, aun cuando su aplicación en esta enfermedad continúa bajo estudio. En este sentido, la farmacogenética es una herramienta prometedora para la identificación de nuevos tratamientos farmacológicos más eficaces y seguros para la EA, así como para apoyar el desarrollo de los fármacos que se encuentren actualmente en investigación.
Ninguno de los fármacos desarrollados para detener la producción, agregación o promover la eliminación del péptido Aβ ha demostrado ser un tratamiento eficaz en los ensayos clínicos de fase III realizados hasta la fecha, por lo que será necesario investigar nuevas dianas terapéuticas principalmente en la etapa preclínica de la enfermedad.
La identificación de biomarcadores farmacogenéticos que puedan predecir la respuesta a fármacos inhibidores de acetilcolinesterasa y la memantina contribuirían a tomar mejores decisiones en el tratamiento de la EA, en un contexto tan complejo como lo es el envejecimiento caracterizado por una politerapia y comorbilidad, y en el que la presencia de efectos pleiotrópicos puede jugar un rol determinante.
FinanciaciónRealizada por el Consejo Nacional de Ciencia y Tecnología (México). Este artículo de revisión se escribió como parte del Proyecto de Desarrollo Científico para Atender Problemas Nacionales 2016 del Consejo Nacional de Ciencia y Tecnología (N.° propuesta 3099), la estancia posdoctoral financiada por el Consejo Nacional de Ciencia y Tecnología para el doctor Tirso Zúñiga Santamaría y la beca otorgada a Ingrid Fricke-Galindo (CONACYT #369708) para la obtención del grado de doctora.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

